
GENOME BIOLOGY:使用大规模并行拼接分析对拼接变体预测算法进行基准测试
2023-12-25 Jenny Ou MedSci原创 发表于上海

SpliceAI和Pangolin在测试的预测器中表现出最佳的整体性能,然而,仍然需要改进拼接效应预测,特别是在外切体中。
剪接是使用初级转录物中编码的序列信息在mRNA成熟过程中去除内含子的过程。破坏剪接的序列变异会加剧许多人类遗传疾病的等位基因谱,据估计,总体而言,多达三分之一的疾病相关单核苷酸变异是剪接破坏性的。拼接破坏性变体(SDV)在基本拼接位点二核苷酸(U2型内含子为GU/AG)最容易识别,孟德尔疾病中有许多例子。SDV也可以发生在几个所谓的侧翼非规范位置,根据一些估计,这些比基本拼接突变多出几倍。
拼接位点图案之外的变体可能具有类似的破坏性,但识别起来更具挑战性。其中一些破坏了拼接增强器或消音器,这些剪接器或消音器是受拼接因子约束的短图案,以刺激或抑制附近的拼接位点,赋予额外的特异性,并提供受监管的替代拼接。这些剪接调节图案很普遍,并通过净化选择来维持,尽管它们通常具有部分冗余,并且可以耐受一些突变。一些特征最好的病例来自遗传疾病,其中典型的例子是SMN1丢失引起的脊柱肌肉萎缩。
由于消除了外源性拼接增强剂(ESE)的固定差异,其几乎相同的paralog SMN2无法在功能上补充其损失。由此产生的外消子7跳过可以被反感寡核苷酸靶向,以充分促进SMN2蛋白的表达,以提供治疗益处。其他案例包括同义变体,作为一个类别,这些变体可能会被忽视,并可能破坏现有的剪接调节元素或引入新的元素,如ATP6PA2相关X-linked parkinsonism。系统地划定拼接调节元素及其同源因素,并定义它们用于塑造拼接模式的语法或“拼接代码”,一直是分子和计算生物学的长期挑战。
来自患者标本的RNA分析可以为拼接破坏性变体提供强有力的证据,并将其纳入临床基因测试可以提高诊断产量。然而,对受影响基因的预先了解对于有针对性的RT-PCR分析是必要的,而基于RNA-seq的测试尚未广泛存在,两者都依赖于血液或其他临床可访问的组织中的充分表达进行检测。
因此,在基因测试期间仍然需要对SDV进行可靠的硅预测,并为此开发了各种算法。例如,S-Cap和SQUIRLS实现了使用特征的分类器,如拼接点的主题模型、拼接调节元素的kmer分数和进化序列保存,并接受关于良性和致病临床变体集的培训。
最近的许多算法使用深度学习直接从初级序列预测拼接点似然;然后可以通过比较野生型和突变序列的预测来检测SDV。SpliceAI和Pangolin不使用临床变体集进行训练,而是使用基因模型注释,根据每个基因组位置在已知转录物中是否作为接受者或供体进行标记为真/假。SPANR使用主序列使用RNA-seq测量提供的训练数据来预测百分比拼接(PSI)测量。
HAL采取独特的方法,对随机序列库及其实验观察到的拼接模式进行训练,而MMSplice将HAL的训练数据与从主序列和在注释拼接部位和临床变体上训练的附加模块相结合。最后,ConSpliceML是一个元分类器,它将SQUIRLS和SpliceAI分数与基于人群的约束指标相结合,该指标测量了人口数据库中明显健康成年人中预测的拼接破坏性变体的区域消耗。虽然这些大多是通用短变体预测器,但其他工具是专门为更专业的上下文构建的,例如同义变体和深层内电子变体。
鉴于拼接预测器的激增及其在变体解释中的实用性,了解其性能特征很重要。之前的比较表明,总体而言,SpliceAI代表了最先进的技术,其他几种算法,包括MMSplice、SQUIRLS和ConSpliceML,表现出有竞争力或在某些情况下更好的性能。然而,迄今为止的基准测试工作主要依赖于精选的临床变体,这些变体对规范拼接位点突变非常丰富,可能是由于其分类相对容易。
这就留下了一个问题,即这些工具的性能可以推广到什么程度,以及某些工具在特定情况下是否可能出类拔萃(例如,外显式神秘拼接激活突变)。另一个挑战是,其中一些工具的训练数据可能与基准验证集部分重叠,如果不仔细识别和删除重叠的变体,这些测试可能会存在循环性。
大规模并行剪切分析(MPSA)提供了一个与临床和人群变异集完全正交的对剪切效应预测器进行基准测试的机会。MPSA以汇总的方式测量数千种变体的拼接效果:细胞通过克隆成迷你基因结构的变体库进行转染,该微基因结构具有深RNA测序,作为变体拼接结果的定量读数。MPSA有几种不同的口味:宽广的MPSA屏幕评估许多外切体,并测量每个的一个或多个变体的影响,而饱和屏幕专注于单个外切体或图案,并测量每个目标中每个可能点变体的影响。
两个广泛的MPSA数据集,Vex-seq和MaPSy,最近被用作对拼接效应预测器进行基准测试,作为基因组解释(CAGI)竞争的关键评估的一部分,另一个,MFASS[14]已被用于验证最近的元预测器。然而,使用广泛的MPSA进行基准测试的一个局限性是,它们可能反映了外消声器的整体特性,同时缺乏评估其中不同变体的更精细分辨率。例如,算法可以通过预测具有弱剪接位点或进化保守序列的外显子中的SDV来表现良好,同时未能区分每个变体中真正的破坏性和中性变体。

2023年12月21日发表在GENOME BIOLOGY的文章,利用饱和MPSA作为互补的、高分辨率的基准数据来源来评估八个最近和广泛使用的拼接预测器。使用深度学习来模拟拼接影响的算法,使用广泛的侧翼序列上下文,SpliceAI和Pangolin,始终显示与测量的拼接效果的最高一致性,而其他工具在特定外显子或变体类型上表现良好。即使对于性能最好的工具,预测也与外来变体和内电子变体的测量效果不一致,这表明未来算法的一个关键改进领域。
研究人员对八种广泛使用的拼接效应预测算法进行基准测试,利用大规模并行拼接分析(MPSA)作为实验确定的地面真相的来源。MPSA同时分析许多变体来提名候选SDV。本文将实验测量的拼接结果与五个基因中3616个变异的生物信息预测进行了比较。算法与MPSA测量的一致性,以及彼此之间的一致性,在外显性比内显性变体要低,这凸显了识别错误或同义SDV的难度。
研究结果显示,基于基因模型注释训练的基于深度学习的预测器在区分破坏性和中性变体方面实现了最佳的整体性能,并控制整个基因组的呼叫率,SpliceAI和穿甲具有卓越的灵敏度。最后,本文结果强调了在对整个基因组的变体进行评分时的两个实际考虑因素:找到最佳得分截止,以及基因模型注释的差异带来的巨大变异性,建议在面对这些问题时进行最佳拼接效应预测的策略。
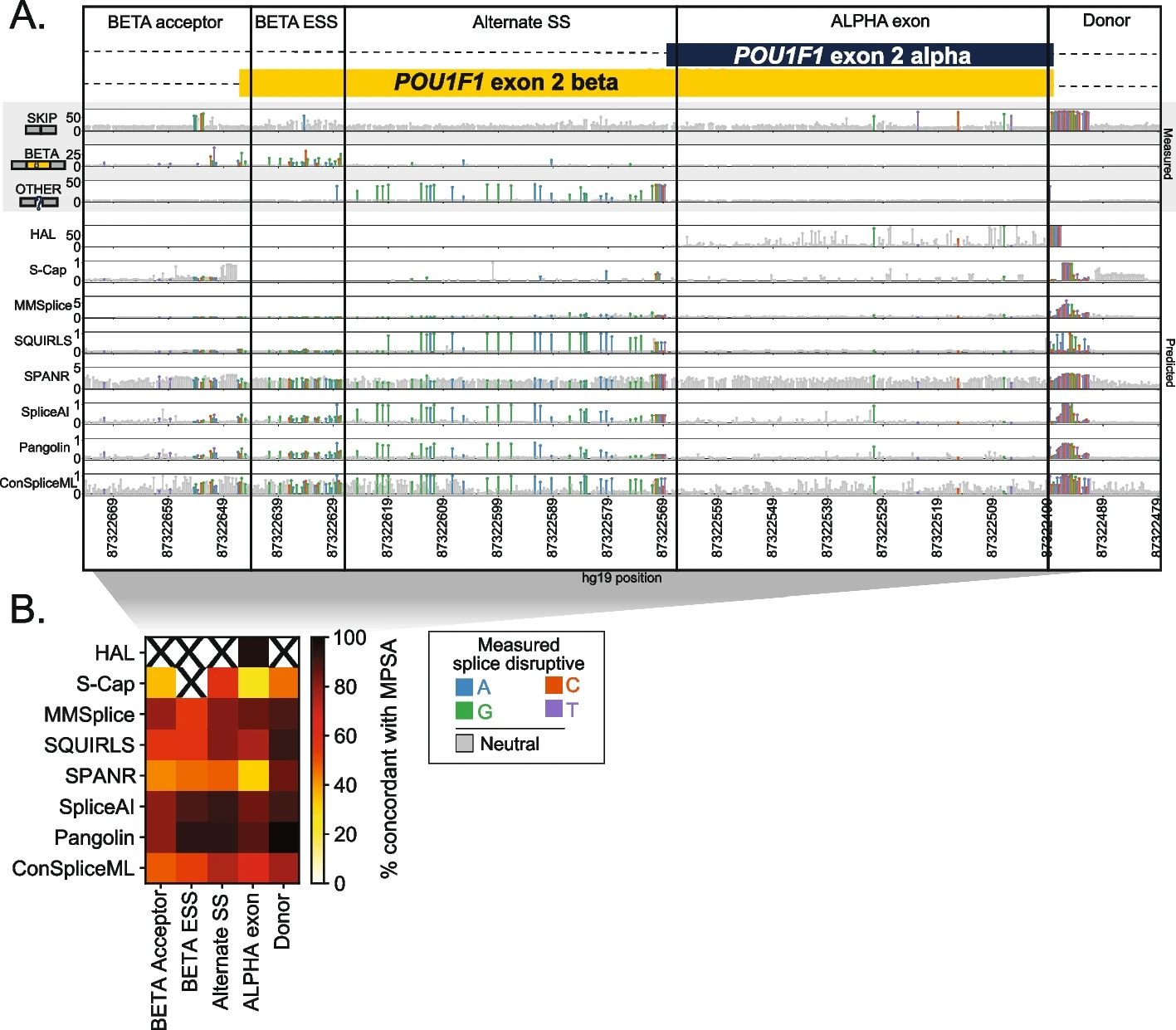
预测因子和实验之间的一致性因基因区域而异
综上所述,饱和MPSA为批判性地评估计算拼接效应预测器的性能提供了机会。本文结果通过测试算法在几个外显子背景下区分许多变体影响的能力,补充了过去使用临床变体和更广泛地针对MPSA的基准测试工作。这种分类任务类似于临床医生在变异解释时面临的任务,因为有许多罕见的变异,即使在容易发生剪接破坏的疾病基因外源中,也不会影响剪接。
本文确定SpliceAI和Pangolin是性能最好的工具,但指出了包括外源变体性能在内的缺点,以及最终用户可能遇到的实际挑战,包括阈值的选择和需要仔细关注基因模型注释。MPSA屏幕的持续增长将为进一步改进拼接效应预测器提供机会,以帮助解释变体的拼接影响。
原始出处
Smith, C., Kitzman, J.O. Benchmarking splice variant prediction algorithms using massively parallel splicing assays. Genome Biol 24, 294 (2023). https://doi.org/10.1186/s13059-023-03144-z
 s13059-023-03144-z.pdf
s13059-023-03144-z.pdf
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#预测算法# #大规模并行拼接分析# #对拼接变体# #基准测试#
65