
重磅综述:一文了解阿尔茨海默症的突触病理机制和靶向突触变性新疗法
2022-12-28 周科 “神经周K”公众号 发表于上海

在AD的所有病理变化中,突触丢失与认知功能下降的关系最为密切。越来越多证据表明AD是病理蛋白、突触和胶质细胞间复杂相互作用的病理结果。
阿尔茨海默病(AD)是最常见的神经退行性疾病,目前仍无有效的治疗方法。AD的病理特征是淀粉样蛋白-β(Aβ)斑块和磷酸化tau神经原纤维缠结沉积导致的进行性神经元和突触丢失。在AD的所有病理变化中,突触丢失与认知功能下降的关系最为密切。越来越多证据表明AD是病理蛋白、突触和胶质细胞间复杂相互作用的病理结果。
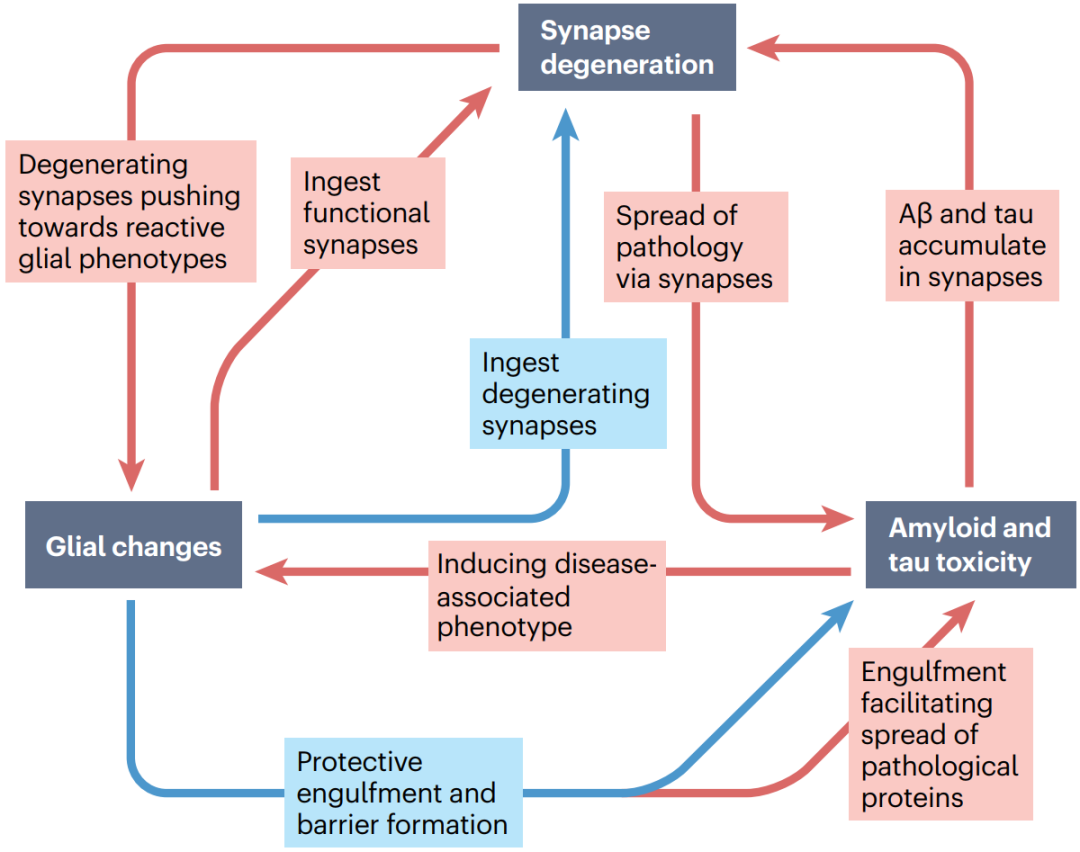
2022年12月13日英国爱丁堡大学脑科学研究中心的Tara L. Spires-Jones在Nature Reviews Neurology期刊发表的综述总结了AD突触病理机制的研究进展,重点介绍了其中病理蛋白、突触和胶质细胞间的相互作用(图1),并讨论了靶向突触变性的AD新疗法和生物标志物研究的发展现状及前景。
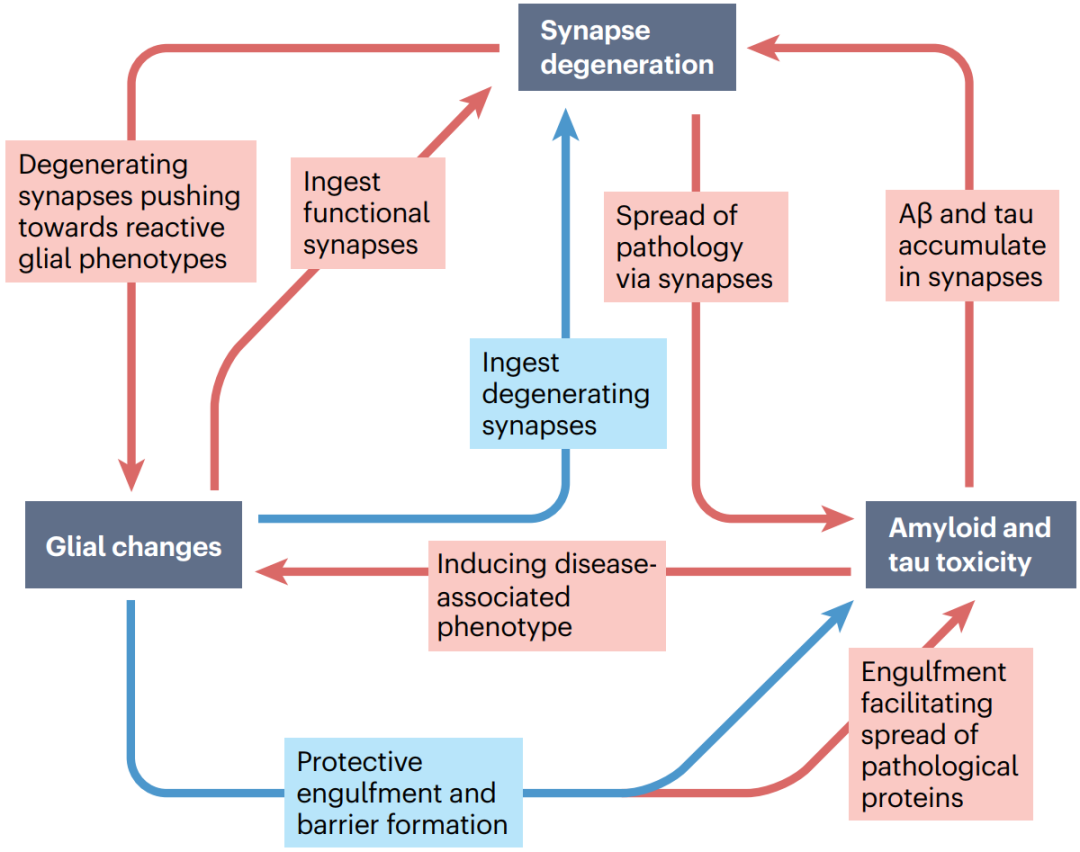
图1. 病理蛋白-胶质细胞-突触变性的相互作用概括
1. Aβ和tau蛋白在突触变性中的作用
除Aβ和tau组成的斑块和缠结外,大量证据表明Aβ和tau的可溶性低聚物可在大脑不同区域传播,直接导致突触功能障碍和丢失(图2)。AD中,Aβ和tau蛋白低聚物分别积聚在突触大量丢失区域的胞外和突触胞质中,清除这些蛋白可改善突触功能。Aβ和tau蛋白病理相互作用,Aβ为tau的突触聚集创造环境,而敲除tau蛋白可防止Aβ诱导的突触缺陷。

图2. Aβ和tau在AD突触变性中的作用机制
Aβ的突触毒性
Aβ的突触毒性机制尚未阐明。研究发现Aβ低聚物可靶向并结合突触,与多种突触受体相互作用。
细胞朊蛋白(PrPC)被认为是Aβ低聚物受体。通过与PrPC结合,Aβ与代谢性谷氨酸受体5 (mGluR5)相互作用并诱导其异常聚集,过度调动细胞内钙储存,破坏突触蛋白翻译。Aβ-PrPC结合也可导致Fyn激活,引起tau蛋白过度磷酸化。因此,Aβ-PrPC反应被认为是AD潜在治疗靶点,但以其为靶点的塞卡替尼(AZD0530)在临床试验中未获得理想结果。
NMDARs通过诱导过量钙内流介导Aβ的突触毒性,Aβ低聚物以NMDARs依赖的方式抑制LTP和降低树突棘密度,机械敏感通道在其中发挥关键作用。Aβ还可诱导突触外NMDAR激活,引起cAMP反应元件结合蛋白(CREB)关闭,导致突触蛋白减少和树突棘丢失。然而,NMDAR拮抗剂美金刚在AD临床试验中的疗效有限。
Aβ还可与其它神经元受体相互作用。例如,Aβ与ephrin B型受体2(EphB2)结合诱导其降解,引起LTP和记忆损伤;与神经元特异性Na+/K+- ATP酶α3亚基(NAKα3)结合诱导线粒体功能障碍;与卷曲受体结合损伤突触可塑性;与树突胰岛素受体结合诱导其表达下调,抑制突触生长和存活。此外,一些分子如ADAM10可改善Aβ诱导的突触变性,其通过促进神经保护性sAPPα储备和限制突触内的内源性A β合成在突触后末梢的形成和稳定中发挥重要作用。
2. tau的突触毒性
通常情况下,tau蛋白通过与细胞内突触蛋白作用产生突触毒性。大量研究表明,tau蛋白可直接与synaptophysin、synapsin 1、synaptotagmin和synaptogyrin 3等多种突触蛋白相互作用,参与突触前囊泡池损伤、突触前蛋白水平降低、caspases激活和线粒体功能障碍。此外,Tau蛋白在AD中可以错定位到突触后树突,与Fyn激酶作用而破坏树突棘形态并导致棘丢失,或促进微管蛋白酪氨酸连接酶样6 (TTLL6)易位而导致微管破裂、突触线粒体募集中断和棘丢失。通过NMDARs介导的Aβ毒性依赖于下游tau和Fyn激酶之间的相互作用,这个组合在AD中被称为“毒性三联体(toxic triad)”。
3. 钙失调
可溶性Aβ和tau都可以通过破坏钙稳态而损伤突触功能。在突触后,Aβ低聚物可直接与突触质膜相互作用、或通过过度激活NMDARs驱动钙内流,进而激活钙调神经磷酸酶,导致树突棘的物理崩溃。在突触前,A β诱导的钙内流导致静息池中囊泡回收途径受损及囊泡过度积累。此外,A β介导的钙内流通过NMDARs导致tau过度磷酸化,促进其树突错定位。
4. 突触线粒体功能障碍
功能性突触线粒体损伤是Aβ和tau蛋白的共同病理结果。AD中,突触前线粒体数量减少,且Aβ斑块周围突触线粒体形态被破坏,Aβ低聚物可直接与线粒体作用,促进氧自由基产生、细胞凋亡和线粒体结构破坏。AD中的Tau病理可阻止线粒体进入突触,抑制突触前囊泡释放。此外,磷酸化的tau或Aβ与动力相关蛋白1结合会导致线粒体碎片过度产生。
5. Aβ病理扩散
AD的临床前和前驱期以默认模式网络(DMN)异常亢进为特征,DMN的活动涉及颞叶、顶叶、额叶和小脑,这些区域在疾病早期可发现Aβ斑块积聚。DMN中异常增加的突触传递可能加剧Aβ释放,促进其积聚并从该网络扩散到海马及其外连接结构。目前,Aβ通过神经环路跨突触传播的直接证据仍然缺乏。相比之下,大量研究证实tau蛋白可通过突触连接进行传播。
6. 胶质细胞
AD蛋白病理可推动小胶质细胞和星形胶质细胞病变,加剧突触变性。在AD中,补体系统以非特异性的方式标记健康和退化的突触,诱导胶质细胞介导的过度突触消除,导致进一步的突触功能障碍和认知能力下降(图3)。此外,胶质细胞来源的炎性细胞因子也可促进AD中的突触和神经毒性。多项证据表明抑制小胶质细胞的突触消除可改善突触和认知功能障碍,减少小胶质细胞数量可能是调节AD病理的有效策略。

图3. AD中胶质细胞-突触相互作用
2.AD新治疗靶点的临床试验
新的AD疗法可通过诱导突触健康的自主调节机制或通过调节胶质细胞的活性直接或间接作用于突触。然而,由于AD突触变性的机制研究过度依赖于动物模型,新疗法的开发受到限制。有证据表明,99%在临床前模型中有效的药物在临床试验中效果不佳。
目前,所有被批准的AD治疗药物都以突触受体(乙酰胆碱或NMDA受体)为靶点,且疗效有限。正在进行的AD疗法的临床试验中,一部分将突触完整性的检测作为主要终点,例如,已在II期试验取得积极结果的年轻血浆成分GRF6019(Alkahest/Grifols Biologicals);另一部分旨在保持AD患者的突触完整性(作为次要终点),所用策略包括预防Aβ诱导的突触毒性(CT1812,Cognition Therapeutics;BPN14770,Shionogi Pharma/Tetra Therapeutics)、促进营养因子(苔藓虫素1)或直接刺激突触环路(GENUS196, Cognito Therapeutics)。此外,大量临床试验正在测试针对病理性Aβ或tau沉积的治疗方法,通过减少直接突触毒性和减少胶质细胞增生治疗突触变性。
3.突触的生物标志物
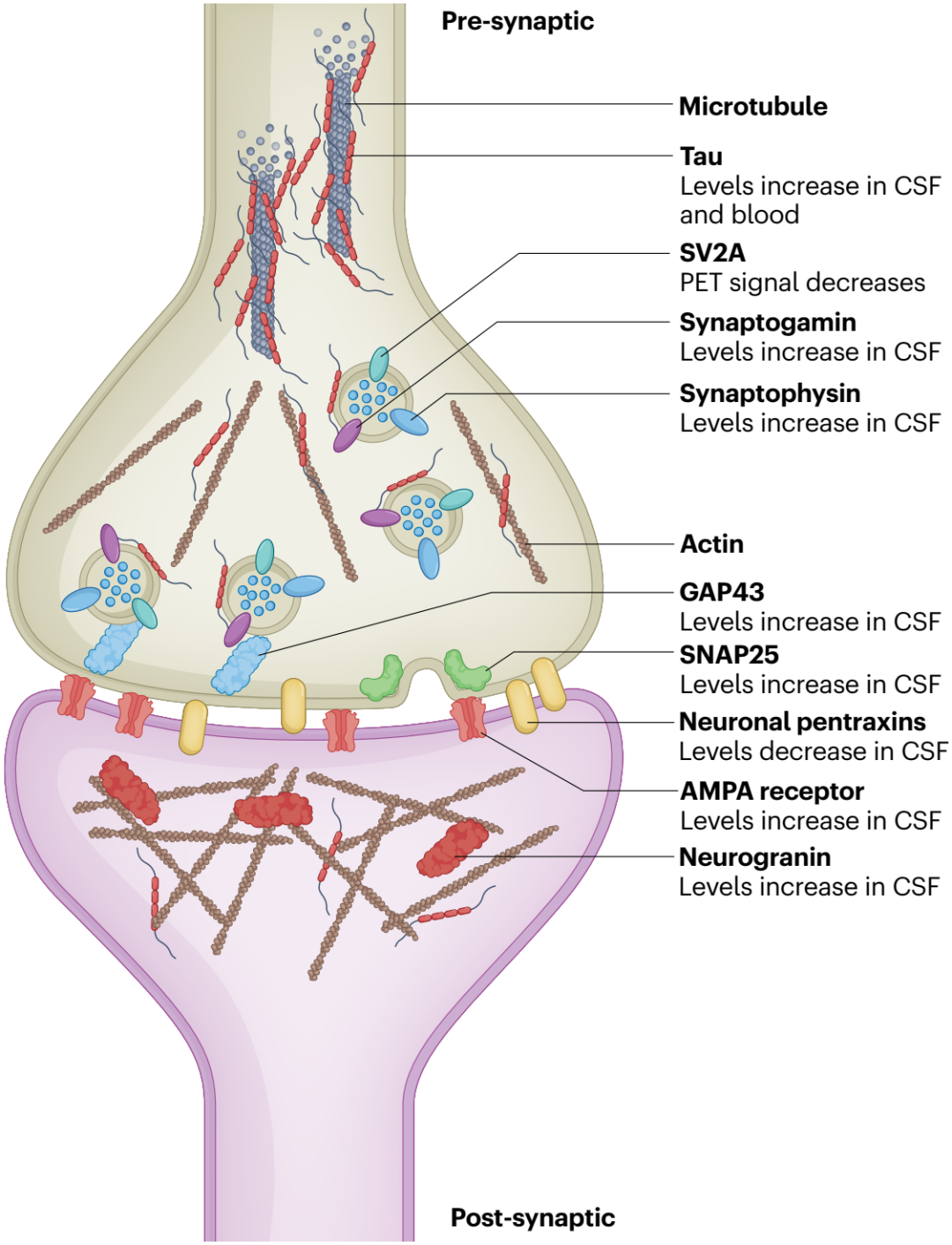
AD中突触变性的研究推动了其生物标志物的发展,目前AD突触生物标志物以脑脊液(CSF)中的突触蛋白为主。例如,AD患者CSF中增加的神经颗粒蛋白、SNAP25、synaptotagmin、synaptophysin、RAB3A、GAP43和AMPA受体亚单位等突触前/后蛋白。其中,神经颗粒蛋白的增加特定于AD,并与未来的认知能力下降、葡萄糖代谢和脑萎缩相关(图4)。
结合突触前囊泡(SV2A)的PET配体被用来观察活体AD患者中突触丢失模式,例如,内嗅皮层SV2A PET信号与tau PET信号呈负相关,提示内嗅皮层投射神经元中tau病变导致下游脑区突触丢失。SV2A PET配体有望准确地在AD患者体内反映突触丢失,为临床试验中新疗法的评估提供助力。
总结
AD突触变性的机制包括Aβ和tau的可溶性低聚物或纤维沉积引起的突触毒性和胶质细胞介导的过度突触消除。CSF中突触蛋白检测和活体突触PET成像可能有助于AD早期诊断,并解释机体对衰老认知损伤的抵抗机制。
虽然目前已有大量临床试验聚焦于AD中胶质细胞功能的调节,胶质细胞对病理性突触消除作用尚需更多关注。AD中突触变性机制的高复杂性决定单一疗法很难实现,未来AD治疗可能针对疾病的临床前和前驱期,降低毒性蛋白在突触的聚集水平并减缓进行性突触变性。
原始出处:
Tzioras, M., McGeachan, R.I., Durrant, C.S. et al. Synaptic degeneration in Alzheimer disease. Nat Rev Neurol 19, 19–38 (2023). https://doi.org/10.1038/s41582-022-00749-z.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









老年痴呆病人越来越多啊
71