Nature Neurosci 全面综述Aβ与tau在阿尔茨海默病中的协同作用
2023-02-17 brainnew神内神外 brainnew神内神外 发表于安徽省

阿尔茨海默病(AD)患者的脑内的重要病理指征是同时存在胞外β淀粉样(Aβ)斑块和细胞内tau蛋白积累导致的神经纤维缠结。
阿尔茨海默病(AD)患者的脑内的重要病理指征是同时存在胞外β淀粉样(Aβ)斑块和细胞内tau蛋白积累导致的神经纤维缠结。主流观点认为,AD发病机制是Aβ的增多加速了疾病的进程,并“触发”了包括tau病理学和神经退行性变的有害级联反应;并基于假定“Aβ和tau在非特定条件下是相互独立的”。然而,现在越来越多的证据表明,Aβ和tau存在协同作用。这不仅解释了针对Aβ临床试验的阴性结果,同时也提示,仅针对tau的临床试验可能需要重新考虑。
因此,伦敦大学痴呆症研究所的Marc Aurel Busche和麻省总医院神经退行性疾病研究所的 Bradley T. Hyman联手在近期Nature Neuroscience上发表综述Synergy between amyloid-β and tau in Alzheimer’sdisease,这篇综述从大量的人类和疾病模型数据中,找到了与复杂的Aβ–tau相互作用有关的最新证据基础,并强调了其对于阐明AD发病机制和设计下一代AD临床治疗试验的重要意义。
 1Aβ和tau:是因果关系,还是致病相互作用?
1Aβ和tau:是因果关系,还是致病相互作用?
目前的疾病模型表明,无论是哪种形式的Aβ(斑块、非纤维、可溶性、寡聚体),都会引发病理生理级联反应,导致tau蛋白错误折叠和组装,扩散到整个皮层,最终导致神经系统衰竭、神经退化和认知能力下降,这看起来似乎一种“线性”过程。然而,AD显然存在一种“非线性”的发展,有“Aβ依赖”(影响发病年龄)和“Aβ非依赖”(影响进展率)阶段。所以,临床上一旦出现症状,抗Aβ治疗对改变进展率的益处微乎其微。
动物实验的结果往往看起来好像tau的过度磷酸化是Aβ诱导的下游变化,但临床上并非如此,提示Aβ和tau之间存在有复杂的相互关系,整个疾病过程并没有前后因果,或许对AD最有效的治疗方法是联合抗Aβ沉积和抗tau导致的神经纤维缠结。
2临床为我们带来的提示
PET结果显示,Aβ斑块出现在临床症状前10-20年,tau聚集通常出现在60岁后,当然还有在孩童时期就出现tau病理的案例,所以Aβ和tau说不清先后关系,或许是一种协同的存在。脑脊液生物标志物的研究也证明了Aβ和tau在预测记忆衰退方面的协同关系,只有脑脊液中发现Aβ沉积时,tau的磷酸化水平才和认知能力相关。还有研究发现,AD大脑中的糖代谢变化和脑萎缩是由Aβ和tau结合后导致的。最近一项将tau–Aβ-PET与人类基因表达数据相结合的研究揭示了tau和Aβ进程与脂质代谢基因相关,从而为Aβ和tau之间的协同关系提供了进一步证据。
3AD模型的机制研究
细胞实验证实了Aβ和tau之间的相互作用,比如在表达tau的细胞中添加Aβ后,tau就逐渐开始聚集。这种现象在多种转基因AD小鼠模型中也得到了验证。反之,诱导tau的聚集会导致Aβ斑块的增加。目前还不清楚这种现象的潜在机制是什么,只是猜测可能影响了小胶质细胞,从而产生了细胞的神经退行性病变。现有的动物模型过表达的突变体产生的效应和人体内的不同,同时,不同的转基因鼠的启动子也不同,所以可能存在基因时空表达差异的影响。
4Aβ–tau协同作用对神经系统功能的影响
突触的兴奋性受到Aβ和内源性tau的影响,甚至tau对神经元活动的影响是独立于其缠结状态的。作者实验室最近的两项研究(图1)通过钙离子成像技术发现Aβ依赖的神经元过度兴奋性能够被tau阻断,这可以说是在神经回路角度的拮抗作用,然而进一步的实验解释了在低兴奋性水平上的协同作用,过表达APP的小鼠中,tau以来的低兴奋性显著加快,此外,在Aβ存在的情况下,tau抑制在减轻神经元低兴奋性表型方面效果较差。
 图1:Aβ与tau的相互作用影响神经回路
图1:Aβ与tau的相互作用影响神经回路
5Aβ-tau协同作用的预测机制
我们都知道了Aβ与tau的直接相互作用,作者把新意和关注点放到了小胶质细胞,小胶质细胞表达的基因TREM2已成为AD的明星风险因素。小胶质细胞活化是AD的一个重要精神病理特征,早期可溶性Aβ触发了小胶质细胞的活化,使其释放炎性因子,同样,tau的过表达也驱动了小胶质细胞的激活,这些激活甚至出现在神经纤维缠结之前。这种现象在多种转基因模型和细胞模型中都得到了验证,可以说,小胶质细胞是Aβ与tau协同作用的关键中介(图2)。
小胶质细胞在Aβ存在下促进tau活性和扩散的机制简言之,就是活化的小胶质细胞可以吸收tau,处理后以更具生物活性的形式释放它。神经元吸收释放的tau,并反过来将tau释放到神经鞘膜中。神经元活动可通过多种机制增强,包括Aβ-介导的谷氨酸再摄取阻滞、突触功能受损或血脑屏障(BBB)破裂,导致神经毒性产物,例如白蛋白的外渗和星形胶质细胞TGF-β信号的激活。
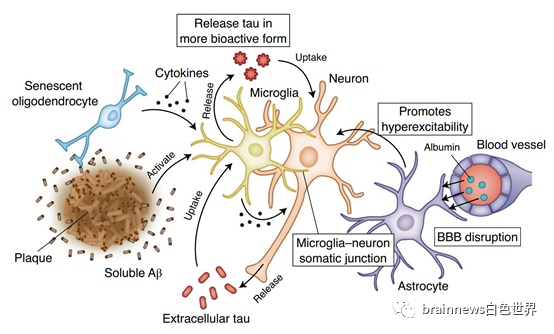 图2:小胶质细胞可能是Aβ-tau协同作用的关键中介
图2:小胶质细胞可能是Aβ-tau协同作用的关键中介
另外,神经元活动在AD中还构成了恶性循环,即Aβ提高了神经元兴奋性,过度活跃的神经元增加了Aβ的沉积和tau的分泌。来自小鼠的数据表明Aβ协同增强了神经元和突触的tau依赖性下调(图3a)。有趣的是,尽管抑制tau能阻止这些基因的进一步下调,但它并未恢复小鼠的正常基因表达水平(图3b)。
Aβ介导的谷氨酸再摄取阻断导致动作电位升高与通过L型电压门控Ca2+通道(L-VGCC)的Ca2+内流有关。Ca2+升高刺激信号级联,包括Ras丝裂原相关蛋白激酶(MAPK)、钙调素依赖性蛋白激酶(CaMK)和钙调神经磷酸酶介导的信号通路的激活,这些信号通路具有多种效应,包括蛋白质的翻译后修饰等(图3c)。
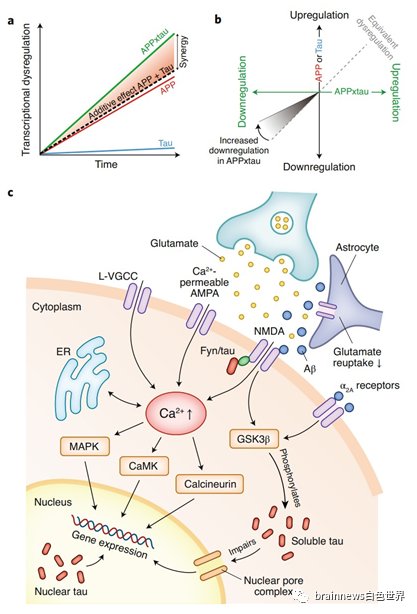 图3: Aβ和tau通路可能在基因表达水平上会聚
图3: Aβ和tau通路可能在基因表达水平上会聚
6总结与展望
总结这篇文章,Marc和Bradley用确凿的实验和临床数据表明,在AD中Aβ和tau之间存在协同作用,这种协同作用不仅表现在整个疾病过程中,而且从根本上推动了疾病的发展。因此,读者们也很容易想到,从Aβ-tau协同作用的角度重新认识AD可能会为未来提供一个重要的且合理的,甚至是成功的治疗方案。
然而,现阶段我们对Aβ-tau协同作用的认识仍处于初级阶段,尚有未解之题,包括不一致的病因或其他关键过程,比如胶质细胞活化如何发挥作用;此外,还需要进一步深入了解包括血管变化和衰老等问题的影响,找到Aβ-tau协同作用的关键机制;需要进行组学分析将模型动物的数据与人的数据联系起来,不断调整和推动临床发展。另外,其他病理因素是否也存在协同作用?这些协同作用又如何影响Aβ和tau之间的协同作用?这些都是对理解像AD这样的复杂且无序的疾病的挑战。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




受教
57