Angelman综合征(天使综合征):症状和体征,病因,流行病学,诊断与治疗
2022-08-17 MedSci原创 MedSci原创

Angelman综合征(AS),又称天使综合征(happy puppet syndrome)或快乐木偶综合征(obsolete),是由母源15号染色体q11-13上的印记基因缺陷导致的一种神经发育性疾
Angelman综合征(AS),又称天使综合征(happy puppet syndrome)或快乐木偶综合征(obsolete),是由母源15号染色体q11-13上的印记基因缺陷导致的一种神经发育性疾病。新生儿通常具有正常表型,发育迟缓最早出现在六个月左右,典型特征一般一岁之后方才显现。患者临床表现为重度精神发育迟滞、小头畸形、共济失调、不合时宜的大笑、癫痫、言语发育障碍、喜欢水和睡眠障碍等。目前,国外报道AS的发病率约为1/15,000-1/20,000。调查研究结果提示,不同人群AS发病率存在明显差异。目前,对于中国人群的AS发病率尚未有系统性的研究。
1965 年,英国医生 Harry Angelman 博士在医学文献中首次描述了 Angelman 综合征。 Angelman 综合征的特征性表现通常在出生时并不明显,并且该疾病的诊断通常在 1 至 4 岁之间进行。
一、一般概述
Angelman 综合征是一种罕见的遗传和神经系统疾病,以严重的发育迟缓和学习障碍为特征;没有或几乎没有讲话;无法协调自主运动(共济失调);颤抖,手臂和腿的动作不平稳,行为模式独特,以快乐的性格和无端的笑声和微笑为特征。尽管患有这种综合症的人可能无法说话,但许多人逐渐学会通过其他方式(例如手势)进行交流。此外,儿童可能有足够的接受语言能力来理解简单的语言交流形式。可能会出现其他症状,包括癫痫发作、睡眠障碍和喂养困难。一些患有天使综合征的孩子可能有独特的面部特征,但大多数面部特征反映了正常的父母特征。 Angelman 综合征是由 UBE3A 基因的缺失或异常表达引起的。
二、症状与体征
Angelman 综合征与多种可能的症状有关。 Angelman 综合征的具体症状因人而异。患有 Angelman 综合征的人不会出现下面讨论的所有症状。例如,一些患有 Angelman 综合征的人可能会癫痫发作,而其他人可能不会。大多数人可能无法说话,而少数人的言语有限。

患有 Angelman 综合征的儿童在达到发育里程碑(发育迟缓)方面会出现延迟,并且有严重的学习障碍。患有天使综合征的儿童也有明显的沟通困难。大多数孩子没有发展出说几句话的能力。孩子们通常可以理解简单的命令。年龄较大的儿童和成人可能能够通过手势和/或使用交流板进行交流。
大多数患有 Angelman 综合征的儿童的早期发现是运动或平衡异常,包括由于无法协调自主运动(共济失调)而导致的急促运动。患有 Angelman 综合征的儿童可能会在手腕和肘部弯曲的情况下举起手臂,并且在走路或兴奋时可能会反复拍打双手。也可能出现躯干肌张力降低(张力减退)、手臂和腿部肌肉张力增加(张力亢进)以及异常夸张或活跃的反射反应(反射亢进)。一些患有 Angelman 综合征的儿童会出现手臂和腿的轻微震颤。这些运动障碍可能在婴儿期早期(大约 6-12 个月大)就很明显。运动里程碑(例如,步行)通常会延迟。在轻微的情况下,儿童可能在 2-3 岁时开始走路。在更严重的情况下,走路可能会明显缓慢、僵硬和生涩。有些孩子可能要到 5-10 岁才能走路。在大约 10% 的病例中,患有 Angelman 综合征的儿童不会在无人帮助的情况下行走。
患有 Angelman 综合征的婴儿和儿童有一种独特的行为模式,其特点是举止愉快,经常且经常不恰当地出现无端、长时间的笑声和微笑。患有 Angelman 综合征的儿童可能容易兴奋、过度运动和过度活跃。他们是积极的探索者,并且经常可能看起来一直在运动。
患有 Angelman 综合征的人可能患有小头畸形,其中头部的周长小于儿童年龄和体重的正常预期。在许多情况下,也可能发生癫痫发作。癫痫发作通常在 1 到 5 岁之间开始,并且通常在青春期得到改善。
与 Angelman 综合征相关的一些发现比上述症状发生的频率要低。在某些情况下,Angelman 综合征患者可能具有独特的面部特征,包括突出的下巴、深陷的眼睛、异常宽大的嘴巴(marcostomia)和突出的舌头、宽齿距和异常平坦的后脑勺(短头畸形) .
由于吮吸能力差,通常在婴儿期可能会出现喂养问题。患有天使综合征的婴儿也可能有吞咽困难。与 Angelman 综合征相关的喂养问题通常并不严重。患有 Angelman 综合征的儿童或成人可能会出现便秘或胃食管反流障碍 (GERD),这种疾病的特征是胃或小肠内容物回流(回流)到连接口腔和胃(食道)的管中。
其他发现包括过度流口水、斜视(斜视)、由于缺乏某些黑色素色素,皮肤、眼睛和头发的颜色(色素减退)缺乏正常。眼睛缺乏色素可能会导致对光敏感(畏光)、快速、不自主的眼球运动(眼球震颤)和视力清晰度下降(视力)。睡眠障碍,例如睡眠需求减少和睡眠/觉醒周期中断或异常(例如,夜间醒来或比正常早起)是 Angelman 综合征儿童的常见表现。患有天使综合症的孩子也可能对水、爱音乐、对闪亮的物体感兴趣。有些孩子可能对热的敏感性增加。随着 Angelman 综合征患儿年龄的增长,脊柱进行性左右弯曲(脊柱侧弯)可能会变得明显。患有 Angelman 综合征的儿童的青春期通常不受影响,并且可以生育。
患有 Angelman 综合征的成年人可能有更明显的面部特征,例如更突出的下颌(下颌前突)。有些人可能会出现角膜异常突出(圆锥角膜)。随着一些人年龄的增长和关节僵硬(挛缩)的发展,活动能力可能会降低。一些年龄较大的儿童和成人可能容易肥胖。
三、病因
E3 泛素蛋白连接酶 (UBE3A) 基因表达的缺乏会导致 Angelman 综合征。该基因位于染色体15区(15q11-q13),含有16个外显子,覆盖的基因组约120kb,编码由865个氨基酸组成的泛素蛋白连接酶,相对分子质量为100,000,其表达具有组织特异性,仅限于脑组织。UBE3A基因属母源性,即来自母亲的UBE3A基因有表达,其表达缺失导致AS,来自父源的UBE3A基因被甲基化而不表达。
可引起Angelman综合征的UBE3A异常包括基因缺失、基因结构改变或基因功能或表达改变。可以破坏 UBE3A 的遗传机制包括染色体缺失、印记错误、父系单亲二体性和 UBE3A 突变。在大约 10% 的情况下,无法确定原因。在大多数 Angelman 综合征病例中,这些遗传变化似乎是随机发生的(偶尔发生),但在大约 3-5% 的情况下它们可以遗传。目前研究表明,5种明确机制会导致AS的发生:
a.母源缺失(70-75%):在UBE3A基因区域母源染色体15q11.2-13关键区域发生4-6Mb的大片段缺失,致此母源UBE3A基因表达缺失;这种删除通常是零星发生的(从头发生)并且不会被遗传。家庭中缺失的复发风险估计为 1-2% 或更低。
b.父源单亲二倍体(1-2%):15号染色体父源UPD,因此缺乏母源的UBE3A基因,父源UBE3A基因不具表达活性;该染色体区域的缺失可能是由于复杂的染色体重排,其中 15 号染色体的一段断裂并移动到另一个染色体位置。具有这种删除机制的人复发的风险更大。
c.病例为印迹缺失(imprinting defect)(3%):母源15q11.2-13区域异常印记状态,存在由 DNA 甲基化错误引起的遗传印记缺陷引起UBE3A基因表达异常;其中,又有大约其中的 20% 的病例中,这是由印记中心内的 DNA 缺失引起的;其余 80% 的病例是由基因印记中的未知或未知缺陷引起的。由于具有 DNA 缺失的印记缺陷,Angelman 综合征复发的风险可能高达 50%。
d.单亲二倍体。大约 2-5% 的 Angelman 综合征病例是由单亲二体性引起的,这是一种异常情况,即一个人从父母一方接收染色体的两个副本,而不是从父母双方接收一个。在 Angelman 综合征中,15 号染色体的两个副本都可以从父亲那里获得(父系单亲二体性)。结果,该区域只有父系表达的基因,因此 UBE3A 在大脑中根本不表达,因为它通常只在母系衍生的染色体上表达。单亲二体性复发的风险低于 1%。
e. UBE3A突变导致泛素化异常。在 10-20% 的 Angelman 综合征患者中检测到 UBE3A 内的异常变化(突变)。该基因功能丧失导致 Angelman 综合征的所有主要临床特征。 UBE3A 包含创建(编码)泛素连接酶蛋白的说明。这种蛋白质标记其他蛋白质,以便身体能够降解目标蛋白质,这一过程称为泛素化。由于 UBE3A 基因的突变,Angelman 综合征复发的风险可能高达 50%。
这5种机制都会直接或间接引起UBE3A基因低表达或不表达;10-15%病例致病性未知。
一些有 Angelman 综合征症状的个体没有可识别的 15 号染色体异常。该组中的一些个体可能患有不同于 Angelman 综合征的疾病,但其他人可能有未检测到的 UBE3A 基因突变或另一种尚未检测到的突变- 被识别的基因,也可能导致或模仿天使综合征。
Angelman综合征与Prader-Willi 综合征的致病区段相同,都为15q11-q13,不同的是Angelman综合征由母源15q11-q13缺失引起,Prader-Willi 综合征则由父源15q11-q13缺失引起。
四、流行病学
Angelman 综合征影响男性和女性的数量相等。 在一般人群中,Angelman 综合征的患病率估计约为 12,000-20,000 人中的 1 人。 然而,许多病例可能未被诊断出来,因此难以确定该疾病在普通人群中的患病率。
五、鉴别诊断
以下疾病的症状可能与 Angelman 综合征的症状相似。比较可能有助于鉴别诊断。
患有 Angelman 综合征的婴儿通常表现为非特异性的精神运动延迟和/或癫痫发作,因此鉴别诊断通常是广泛的和非特异性的,包括脑瘫、静态脑病或线粒体脑肌病等实体。大多数患有 Angelman 综合征的婴儿会出现颤抖和肢体抽搐,这可能有助于将 Angelman 综合征与这些疾病区分开来。
Mowat-Wilson 综合征可出现提示 Angelman 综合征的发现,包括快乐情感、下颌骨突出、言语减少、小头畸形和便秘。 Mowat-Wilson 综合征通常由 ZEB2 基因的杂合突变引起。
Christianson 综合征是一种 X 连锁疾病,可以模仿 Angelman 综合征,涉及 SLC9A6 基因的突变。临床特征包括明显快乐的性格、严重的认知延迟、共济失调、小头畸形和癫痫症。有些人可能有小脑和脑干萎缩。患有 SLC9A6 疾病的个体可能身体外观较瘦,并且可能在 10 岁之后失去行走能力。
腺苷酸琥珀酸裂合酶缺乏导致琥珀酰嘌呤积累,导致精神运动迟缓、自闭症、肌张力减退和癫痫发作。据报道,运动性失用、严重的言语障碍、过度的笑声、非常快乐的性格、多动、注意力不集中、用嘴说话、发脾气和刻板的动作。
Pitt-Hopkins 综合征的特征是智力障碍、宽口和独特的面部特征,以及间歇性过度换气,随后出现呼吸暂停。它可能与 Angelman 综合征有重叠的特征,例如小头畸形、癫痫发作、共济失调步态和快乐的性格。昼夜换气过度是一些人的显着特征,发生在三岁之后。可以进行 TCF4 基因的突变和缺失筛选。
患有癫痫发作和严重言语障碍的 Angelman 综合征婴儿女孩可能类似于 Rett 综合征,但患有 Angelman 综合征的儿童不会像患有 Rett 综合征的女孩那样有退行性病程,也不会失去有目的的双手使用。患有未确诊 Rett 综合征的大龄女孩也可能具有类似于 Angelman 综合征的特征,导致临床错误诊断为 Angelman 综合征。 MECP2 突变的检测已广泛应用。 (有关这种疾病的更多信息,请在罕见病数据库中选择“Rett”作为您的搜索词。)
有时,出现喂养困难和肌肉张力减退的 Angelman 综合征婴儿被误诊为 Prader-Willi 综合征,因为通过 CGH 或 FISH 检测到的 15q11.2-q13 缺失未被 DNA 甲基化分析证明是母体来源。 详细见:Prader-Willi综合征:症状体征、病因、流行病学、诊断与治疗
其他染色体疾病也可以模仿 Angelman 综合征的一些特征,尤其是 22q13.3 缺失(Phelan-McDermid 综合征)。这种情况可能会出现非畸形的面部特征、缺席或很少说话,以及中度至重度的发育迟缓,有时会出现自闭症谱系中的行为特征。
2q23.1 区域的微缺失可能导致严重的言语延迟、癫痫发作、行为障碍和小头畸形。有些人表现出类似天使综合征的表型。其他微缺失疾病,尤其是通过比较基因组杂交(染色体微阵列分析)检测到的较新的疾病,可能与 Angelman 综合征的某些特征有关。
随着染色体微阵列分析和全外显子组测序在对非特异性智力障碍个体的测试中变得越来越频繁,已经确定了其他类似于 Angelman 综合征的情况,而且这些情况肯定会随着时间的推移而增加。其中一些情况包括: KANSL1 单倍体不足综合征(Koolen-de Vries 综合征); Kleefstra 综合征及其变体; HERC2缺乏综合征;男性 MECP2 重复; MEF2C 综合征和 WAC 相关的智力障碍。
六、诊断
可以根据详细的患者病史、全面的临床评估和特征性发现的识别来诊断 Angelman 综合征。大约 80% 的病例可以通过各种专门的血液测试来确认,例如 DNA 甲基化(检测但不区分染色体缺失、印记中心缺陷和父系单亲二体性)。荧光原位杂交 (FISH) 或最常见的微阵列染色体分析可以检测身体细胞中染色体 15q11-q13 的特征性缺失(见于 70% 的病例)。 Angelman 基因 UBE3A 的突变分析可以检测出大约 10% 的 DNA 甲基化研究呈阴性的 Angelman 综合征个体。 UBE3A 的突变分析可以作为一个单独的测试进行特别排序,但现在更常见的是,UBE3A 突变是通过使用一个完整的外显子组测序面板来识别的,该面板包括一组已知会导致智力缺陷的许多基因,或者当一个人执行一个完整的整体时外显子组测序测试(例如,对大约 20,000 个基因的筛选测试)。
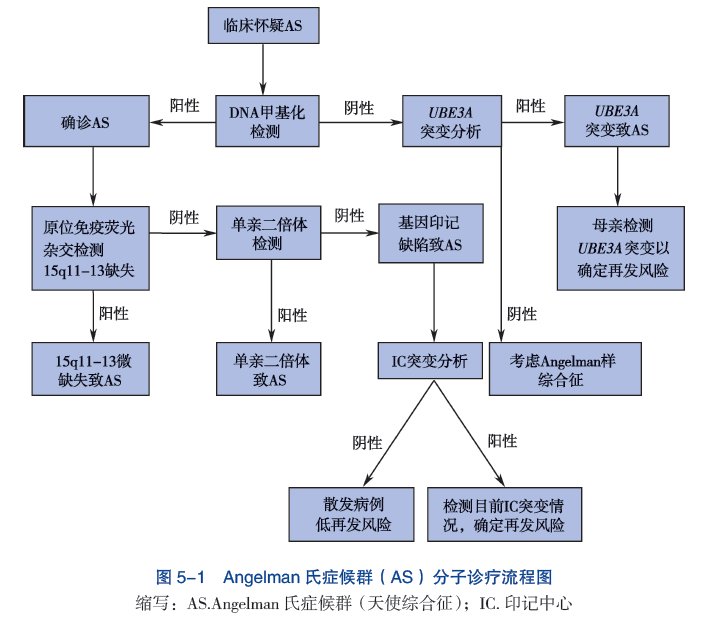
实验室诊断
是确诊的唯一方法,特别是在疾病早期和症状不典型患者的诊断中具有重要作用。
1、DNA甲基化分析
AS患者是由第15号染色体的q11.2-13区域5-7Mb的缺失,单亲二体(UPD)或印记基因缺陷且只有一方未甲基化(异常双亲特定的甲基化印记)。需要注意的是,大多商业化的DNA甲基化分析测试不能够区分AS是由染色体片段缺失,单亲二体(UPD)还是印记基因缺陷(ID)引起的。所以,一些新的方法包括焦磷酸测序,甲基化特异性的多重连接依赖的探针扩增(MS-MLPA),序列为基础的定量甲基化分析(SeQMA)以及拷贝数分析的其他方法作为了解AS的致病机制已投入应用。
2、荧光原位杂交(FISH)与微阵列比较基因组杂交(array CGH)
通过荧光原位杂交(FISH),微阵列比较基因组杂交(array CGH)或其他的缺失检测的方法检测出第15号染色体q11.2-13片段有5-7Mb的缺失的患者检出率率可达到68%(见表1)。
3、单亲二体的分析(UPD)。
利用家族病患先证者和父母双亲的DNA样本进行DNA多态性分析检测单亲二体(UPD),其检出率约为7%。
4、印记中心分析
印记缺失在患者中大约占3%,患者普遍具有异常的DNA甲基化印记但荧光原位杂交(FISH)或微阵列比较基因组杂交(array CGH)分析正常并且无证明显示具单亲二体(UPD)。在印记缺失患者中,大约10-20%的患者由包括AS印记中心(IC)微缺失(6-200kb)所致。这些微缺失可利用多种缺失分析的方法检测出(见表1)。另80%-90%的印记缺失(IDs)患者被认为是在母亲卵子发生过程中或早期的胚胎形成过程中发生了表观遗传突变所致。
5、序列分析
当具有AS临床特征的患者DNA甲基化检测正常时,应考虑对其UBE3A基因序列进行分析。在AS患者中大约有11%具有典型的UBE3A基因突变。有少数AS患者UBE3A基因多外显子或整个基因缺失。这可以利用各种缺失分析的方法检测(见表1)。除此之外,一些微阵列比较基因组杂交技术也可以应用检测这些缺失。
七、标准治疗
此时,Angelman 综合征的治疗是对症和支持性的。几项关于 Angelman 综合征的临床试验正在进行中(见下文),但没有可用的基因疗法或治疗药物。然而,神经科学和基因治疗技术的进步对于提供有意义的治疗和/或治愈该综合征具有巨大的潜力。
Angelman 综合征患者的总体身体健康状况良好,可以提供常规儿科护理,包括常规的儿童免疫接种。
抗癫痫药物(抗惊厥药)对癫痫发作有帮助。通常癫痫发作可以用单一药物充分控制,但在某些情况下癫痫控制可能很困难,需要多种药物。没有一种抗惊厥药物被证明在所有情况下都最有效。睡眠障碍很常见,可能需要行为治疗并遵守严格的睡前程序。有时,镇静药物可能会有所帮助。
喂养困难可以通过改进的母乳喂养方法和特殊乳头等手段来治疗,以帮助吸吮能力差的婴儿。胃食管反流可以通过直立定位和帮助食物通过消化系统运动的药物(运动药物)来治疗。在某些情况下,可能需要通过外科手术收紧连接食道和胃的瓣膜(食道括约肌)。泻药可用于治疗便秘。
脚踝支架/支撑和物理治疗可以帮助实现步行。脊柱侧弯可在 10% 左右发展,可能需要支架或手术矫正。在某些情况下,斜视可能需要手术矫正。
早期干预对于确保患有 Angelman 综合征的儿童发挥其潜力非常重要。可能对患有 Angelman 综合征的儿童有益的特殊服务可能包括特殊的社会支持和其他医疗、社会和/或职业服务。大多数患有 Angelman 综合征的儿童都受益于物理、言语和职业治疗。行为矫正疗法可用于阻止不良行为。使用特殊的通信设备,例如基于计算机图片的系统、发声设备和其他现代技术使用,都有助于 Angelman 综合征患者提供更好的学习和社交交流。
建议对 Angelman 综合征患者的家庭进行遗传咨询。
八、科学研究
2020年2月,GeneTx宣布将开始进行GTX-102治疗Angelman综合征的临床试验,GTX-102是一种实验性反义寡核苷酸药物,正在评估其对Angelman综合征(AS)的安全性和耐受性。这项I/II期开放标签、多剂量研究的目的是评估GTX-102治疗Angelman综合征患儿的安全性、耐受性以及血浆和脑脊液(CSF)浓度。将招募大约20名患儿,这些患者经遗传学确诊为母体UBE3A基因完全缺失。详细见: //m.capotfarm.com/article/show_article.do?id=c69918819419
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Williams CA, Dagli A. Angelman Syndrome. In: Cassidy SB., Allanson JE, eds. Management of Genetic Syndromes. 3rd ed. Hoboken, NJ : Wiley-Blackwell; 2010.
Dan B, ed. Angelman Syndrome. London, UK: Mac Keith Press; 2008.
Dagli A, Buiting K, Williams CA. Molecular and Clinical Aspects of Angelman Syndrome. Mol Syndromol. 2012;2(3-5):100-112.
Williams CA. The behavioral phenotype of the Angelman syndrome. Am J Med Genet C Semin Med Genet. 2012;154C(4):432-7.
Williams CA, Driscoll DJ, Dagli AI. 2010. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385-95.
Calculator S. Augmentative and alternative communication (AAC) and inclusive education for students with the most severe disabilities. Internat J Inclusive Edu. 2007;1-21.
Williams CA, Beaudet AL, Clayton-Smith J, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006:140(5):413-8.
Bruni O, Ferri R, D’Agostino G, Miano S, Roccella M, Elia M. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev. 2004;26(4):233-40.
Clayton-Smith J, Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J Med Genet. 2003;40(2):87-95.
Clayton-Smith J. Angelman syndrome: Evolution of the phenotype in adolescents and adults. Dev Med Child Neurol. 2001;43:467-480.
Laan LA, den Boer AT, Hennekam RC, Renier WO, Brouwer OF. Angelman syndrome in adulthood. Am J Med Genet. 1996;66(3):356-60.
Dagli AI, Mueller J, Williams CA. Angelman Syndrome. 1998 Sep 15 [Updated 2017 Dec 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1144/ Accessed January 25, 2018.
实验性反义寡核苷酸GTX-102治疗Angelman综合征的临床试验即将开始。//m.capotfarm.com/article/show_article.do?id=c69918819419
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#天使人综合征##罕见病#
76
#Angelman综合征#
69
#诊断与治疗#
66
用心了
61
#流行病#
57
#综合征#
65