Nat Mach Intell|基于AI算法预测基因组错义变异的功能影响
2022-12-13 测序中国 测序中国 发表于安徽省

准确预测有害错义变异对于解释基因组序列至关重要。gMVP可以改善临床检测和遗传研究中对错义变异的解释,有望在基因临床检测和新风险基因发现研究中发挥积极作用。
基因组医学研究基础是将个人的基因组特征用于改善医学诊断、预后和治疗。但人体内有数百万个基因变异,其中绝大多数对人的健康影响微乎其微。如何确定变异-疾病关系是基因组医学和精准医疗领域的核心问题之一。
这一问题在改变蛋白质单个氨基酸的错义变异中尤为明显,只有约20-30%的错义变异具有功能影响。对于错义变异来说,一个特定变异是否改变蛋白质功能(导致不良健康状况)是极其不确定的。因此,临床基因检测中的大多数错义变异都被归类为VUS(Variant of Uncertain Significance,意义不明的变异)。
美国哥伦比亚大学系统生物学系和生物医学信息学系副教授沈宇锋博士及其团队前博士后张海仓,开发了一种新方法gMVP来预测哪些错义变异具有潜在功能影响。gMVP使用最新的机器学习技术深度图注意力神经网络模型,来抽取相关特征并预测哪些变异具有功能影响。近期,该研究成果“Predicting Functional Effect of Missense Variants Using Graph Attention Neural Networks”发表在Nature Machine Intelligence上。
gMVP方法使用蛋白质中氨基酸-氨基酸间的共进化信息来确定不同位置间是否在功能上相关,这使得gMVP不仅可以刻画变异所在的一维序列环境,也可以刻画变异所在的蛋白质三维结构环境。研究团队在多个不同的独立数据集上评估了gMVP的性能,涉及的应用包括临床基因测试以及致病新风险基因的发现。在所有测试中,gMVP的表现明显优于其他方法。
沈宇锋博士表示:“已经有多个团队在研究预测错义变异,即使是很小的改进也会带来基因组医学研究的真正变化,但这个问题远未得到解决。任何时候,一旦可以准确地将意义不明变异确定为对患者有害的变异,就可以给患者带来更加有效的干预治疗。”同时,张海仓博士表示:“近几年,AI的快速发展为解决这一重要问题提供了新思路。我们将积累多年的领域知识和AI结合设计了gMVP算法。”
沈宇锋博士表示:“已经有多个团队在研究预测错义变异,即使是很小的改进也会带来基因组医学研究的真正变化,但这个问题远未得到解决。任何时候,一旦可以准确地将意义不明变异确定为对患者有害的变异,就可以给患者带来更加有效的干预治疗。”同时,张海仓博士表示:“近几年,AI的快速发展为解决这一重要问题提供了新思路。我们将积累多年的领域知识和AI结合设计了gMVP算法。”
主要研究内容
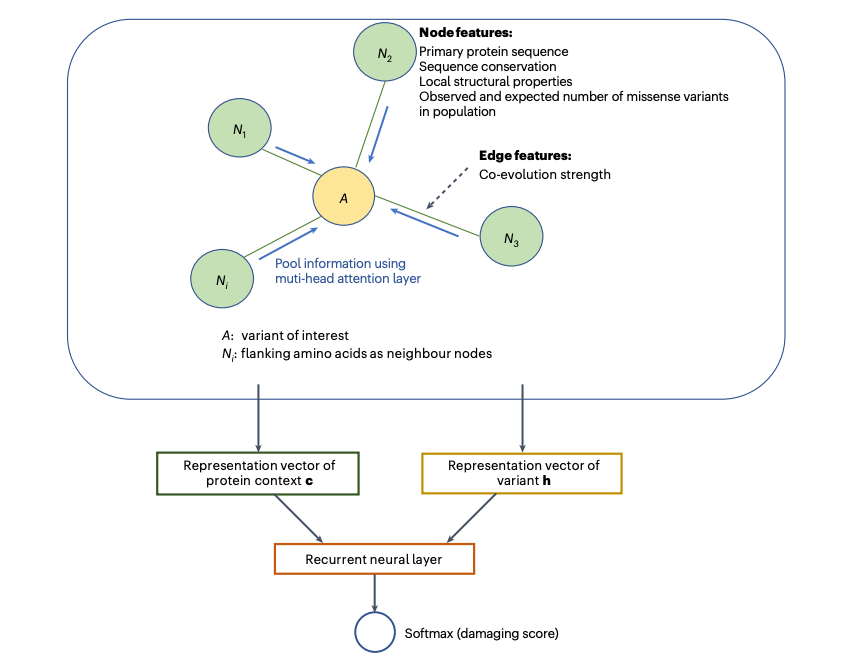
图1.gMVP基于图注意力神经网络模型预测错义变异的功能影响。
gMVP利用图神经网络从蛋白质的序列环境和结构特征中学习变异所在的环境表示 (context representation),并基于该表示预测变异的功能影响。具体地,gMVP用一个图来表示变异以及变异所在的环境。gMVP用氨基酸间的共进化信号作为图上每条边的输入特征,用氨基酸得序列保守性、局部三维结构信息和变异得人群内选择约束作为图上每个点上的特征。共进化信号不仅可以描述变异所在的三维结构微环境,也可也描述氨基酸间得功能相关性。gMVP用多头注意力机制中自动学习每个环境节点的权重,并得到变异及其变异环境的向量表示。然后基于学习到的表示,监督式地学习变异对蛋白质的功能影响。
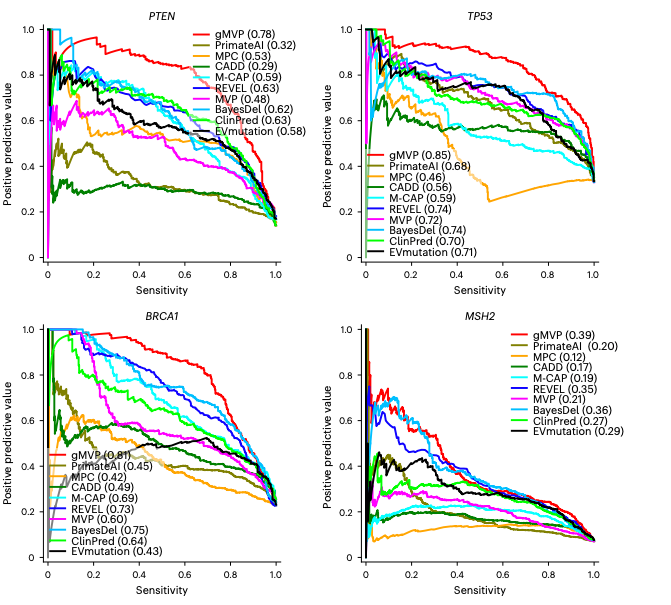
图2.评估gMVP和已发表方法预测癌症风险基因中的有害变异。
gMVP可准确预测癌症风险基因中的有害变异。基于深度突变扫描数据(Deep Mutaional Scanning data),gMVP在4个典型的癌症风险基因上进行测试。gMVP的预测精度显著超过了其他主流算法,包括传统的机器学习算法,例如REVEL、MPC和CADD等,也包括近几年刚发展的基于深度学习的方法,例如PrimateAI和EVmuation等。具体地,gMVP在PTEN、TP53、BRCA1和MSH2上的AUPRCs分别为0.78、0.85、0.81和0.39 ,次优方法的AUPRC分别为0.63、 0.74、0.73 和 0.35。

图3.评估gMVP 和已发表方法区分神经发育障碍患者和对照组的新生突变。
gMVP可以准确区分发育障碍患者和对照人群携带的新生突变(de novo mutations)。在5924个自闭症(ASD)患者,31058个神经发育障碍(NDD)患者和2007个对照组人群的新生突变数据上,gMVP的区分显著性(P-value, Mann–Whitney U test)分别为3×10–9(ASD)和2×10–40(NDD),次优方法分别为38 ×10–6和 28×10–38。
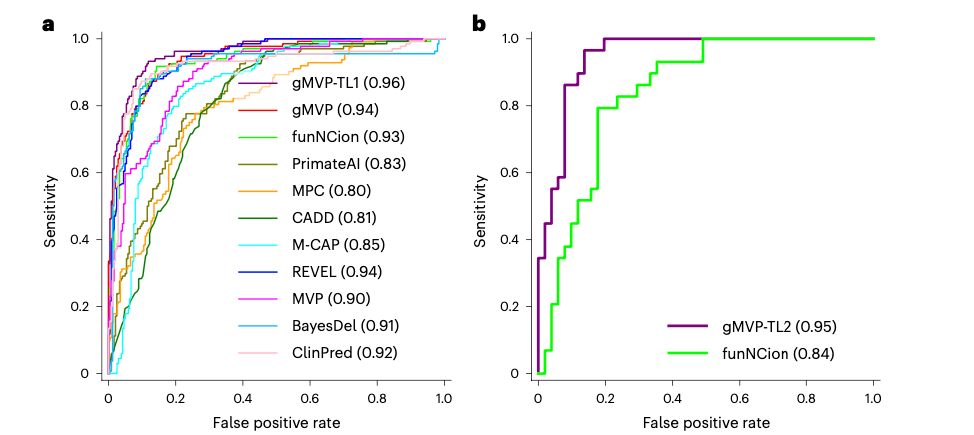 图4.评估 gMVP和其他已发表方法对致病和中性变异进行分类,以及预测离子通道基因中的 GOF和LOF变异。
图4.评估 gMVP和其他已发表方法对致病和中性变异进行分类,以及预测离子通道基因中的 GOF和LOF变异。
gMVP支持迁移学习技术预测获得功能变异(GOF)和丧失功能变异(LOF)。在许多基因中,错义变异的功能影响是复杂的,不能简单地通过二元预测来表示。Heyne等和Mark Daly团队的近期研究了改变钠离子和钙离子通道活性的致病变异,并根据变异携带者的临床表型和电生理学数据区分LOF和GOF变异。gMVP进一步使用已学习的变异环境表示和迁移学习技术对LOF和GOF变异进行分类,而无需额外的人工特征。迁移学习也可以克服LOF/GOF标注数据严重不足的问题。经迁移学习训练的模型gMVP-TL可准确预测LOF/GOF变异,AUROC为0.95,对比方法为0.84。
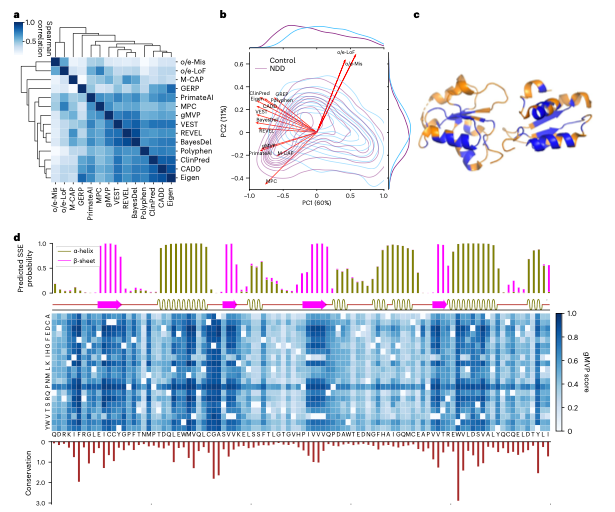 图5.用保守性、蛋白质结构和遗传编码约束解释gMVP预测分数。
图5.用保守性、蛋白质结构和遗传编码约束解释gMVP预测分数。
gMVP的预测分数和模型参数具有很好的可解释性。研究团队以BRCA1基因为例,分别从序列保守性、蛋白质二级结构和三级结构、变异人群内选择约束解释gMVP的预测分数和哪些因素有关。此外,研究团队还从模型学习的氨基酸表示和注意力权重两方面解释gMVP模型。gMVP学习的氨基酸表示可以准确刻画氨基酸的物理化学性质,同时,gMVP学习的注意力权重和氨基酸-氨基酸间的三维距离有很强的相关性。
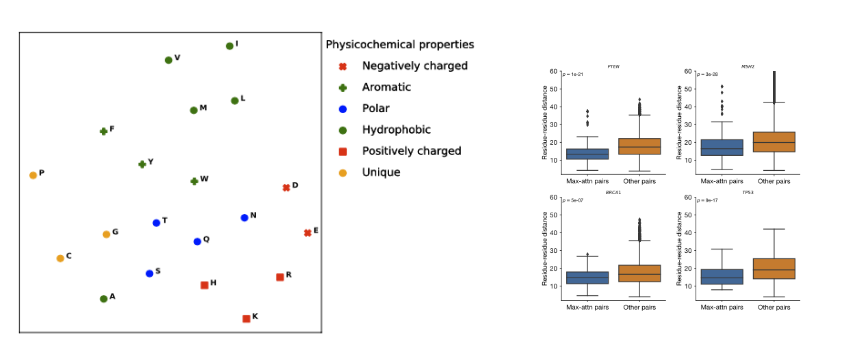
图7.基于氨基酸表示和注意力权重解释gMVP模型。
总 结
准确预测有害错义变异对于解释基因组序列至关重要。gMVP可以改善临床检测和遗传研究中对错义变异的解释,有望在基因临床检测和新风险基因发现研究中发挥积极作用。
主要作者介绍:
沈宇锋(Yufeng Shen),美国哥伦比亚大学系统生物学系和生物医学信息学系,tenured副教授。沈实验室使用基因组学数据和计算方法研究人类生物学和疾病,开发新方法来识别人类疾病的遗传因素,并研究免疫系统的动态适应性。沈实验室的研究领域是计算机科学、统计学、生物学和医学的交叉领域,主要包括计算基因组学、人类遗传学和计算免疫学。
张海仓(Haicang Zhang),之前为哥伦比亚大学沈实验室的博士后,现为中科院计算所副教授、中科大数据研究院特聘研究员。研究领域为AI for Science(生物+AI),主要包括AI辅助的精准医疗,蛋白质结构预测和蛋白质设计,抗体结构预测和抗体设计。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



