
学术交流|蒋镇阳:GTP信号转导串联起代谢、DNA修复和对基因毒性应激的反应
2024-01-05 医悦汇 医悦汇 发表于陕西省

本期文献内容由吉林大学第一医院姜新教授课题组蒋镇阳分享肿瘤治疗相关领域及病理生物学研究领域最新进展,以供交流!
编者按:基因毒性疗法是癌症治疗的里程碑,侵袭性最高的GBM胶质母细胞瘤中,基因毒性的放射治疗和TMZ是标准治疗手段。本期文献内容由吉林大学第一医院姜新教授课题组蒋镇阳分享肿瘤治疗相关领域及病理生物学研究领域最新进展,以供交流!
研究背景
基因毒性疗法是癌症治疗的里程碑。在成人最常见并且侵袭性最高的GBM胶质母细胞瘤中,基因毒性的放射治疗和TMZ是标准治疗手段。DNA损伤反应DDR控制着基因毒性疗法的效率和一般组织毒性。GBM细胞对DNA损伤的快速修复能力导致其对放化疗的耐受性以及肿瘤再发。DDR缺陷的肿瘤对RT等基因毒性疗法更敏感,而DDR缺陷的患者在接受基因毒性疗法后发生更多的一般组织损伤。对DDR生理过程的了解有利于基因毒性疗法相关新药的开发。DDR抑制剂已经尝试在包括GBM的多种肿瘤中与基因毒性疗法联用,然而其诱导一般组织损伤增多的风险限制了其应用。
代谢会调节包括DDR过程在内的细胞表型。作者及其他团队曾找到过细胞代谢过程、DDR过程以及RT或TMZ等基因毒性疗法耐受性之间的关系。代谢物可以从多种不同的机制对细胞表型产生调节,包括调节ROS、驱动生物大分子合成、激活级联信号通路和提供化学能等。代谢物和DDR之间的分子联系仅在少数情况中得到阐明。促进抗氧化合成的代谢物可以通过防止氧化DNA损伤来减轻RT诱导的毒性。吉西他滨和5-FU等药物可消耗脱氧核苷酸,导致细胞周期停滞、复制应激和RT增敏。这两种药物在临床上都与放疗联合用于治疗多种癌症。最近研究发现,肿瘤代谢物可以通过破坏DNA损伤部位的染色质甲基化来削弱HR同源重组修复。这种理论的进步推动了 PARP 抑制剂的临床试验,这些抑制剂选择性地在缺乏同源重组的癌症中有效,并可与基因毒性药物联用于多种癌症。因此,对代谢物如何调节 DDR 的机制理解可以对临床治疗产生影响。
在本研究中,作者明确了癌症和正常组织中核苷酸代谢物与 DDR 之间的治疗相关分子联系。发现GTP是控制DDR的关键核苷酸,而不是其他嘌呤或嘧啶,它通过调节非同源末端连接(NHEJ)来实现。因为只有GTP调节NHEJ,推断GTP依赖性信号转导可能是这种调节的原因,而不是GTP与其他核苷酸共有的作为RNA或DNA前体的作用。使用磷酸化蛋白质组学和各种分子生物学技术及靶向测定,确定了调节 NHEJ 的蛋白质 Abl-interactor 1(Abi-1)上的GTP依赖性去磷酸化事件。Abi-1是一种在DNA修复中没有已知作用的蛋白质,其去磷酸化取决于G蛋白Rac1和蛋白磷酸酶5的活性。调节Rac1和Abi-1活性会影响GBM对基因毒性疗法的敏感性。这些观察结果从GBM扩展到了正常组织,因为GTP补充剂促进了非转化细胞中的Abi-1的去磷酸化,并减轻了RT和基因毒性化疗诱导的正常组织毒性。GTP独立于脱氧核苷酸池调节DNA修复的这种出乎意料的能力对生理学和癌症治疗具有重要意义。
研究结果
结果一:GTP通过NHEJ促进DSB双链断裂修复
在之前的研究中,作者发现是嘌呤而不是嘧啶核苷酸促进GBM在RT后的DSBs修复。这种调节所依赖的核苷酸分子及其机制尚未被阐明。为了明确这些关联,作者首先对患者来源的神经球以及永生化GBM细胞系进行GTP/ATP前体鸟苷/腺苷的补充,并同前所述,利用免疫荧光测量γ-H2AX(S139)灶的形成来估计RT后的DSBs修复效率。发现鸟苷的补充使RT后4小时GBM神经球(图1A,图S1A)和永生化细胞系(图1B,图S1B-D)的γ-H2AX灶数量减少(而腺苷的补充无效)。进一步,在神经球和永生化细胞系中对GTP合成的限速酶IMPDH1/2的沉默延缓了DSBs修复(而ATP合成限速酶ADSS1/2的沉默无效)(图1C,图S1E-F),这种修复的延迟可以被鸟苷补充所解救。用AG2037对两种嘌呤合成(GTP和ATP)的抑制可以延缓RT诱导的DSBs修复,这种现象可以被GTP的补充所挽救(而ATP补充无效)(图1D,图S1G)。至此,作者将GTP定义为GBM模型中RT后DSBs修复的关键核苷酸。
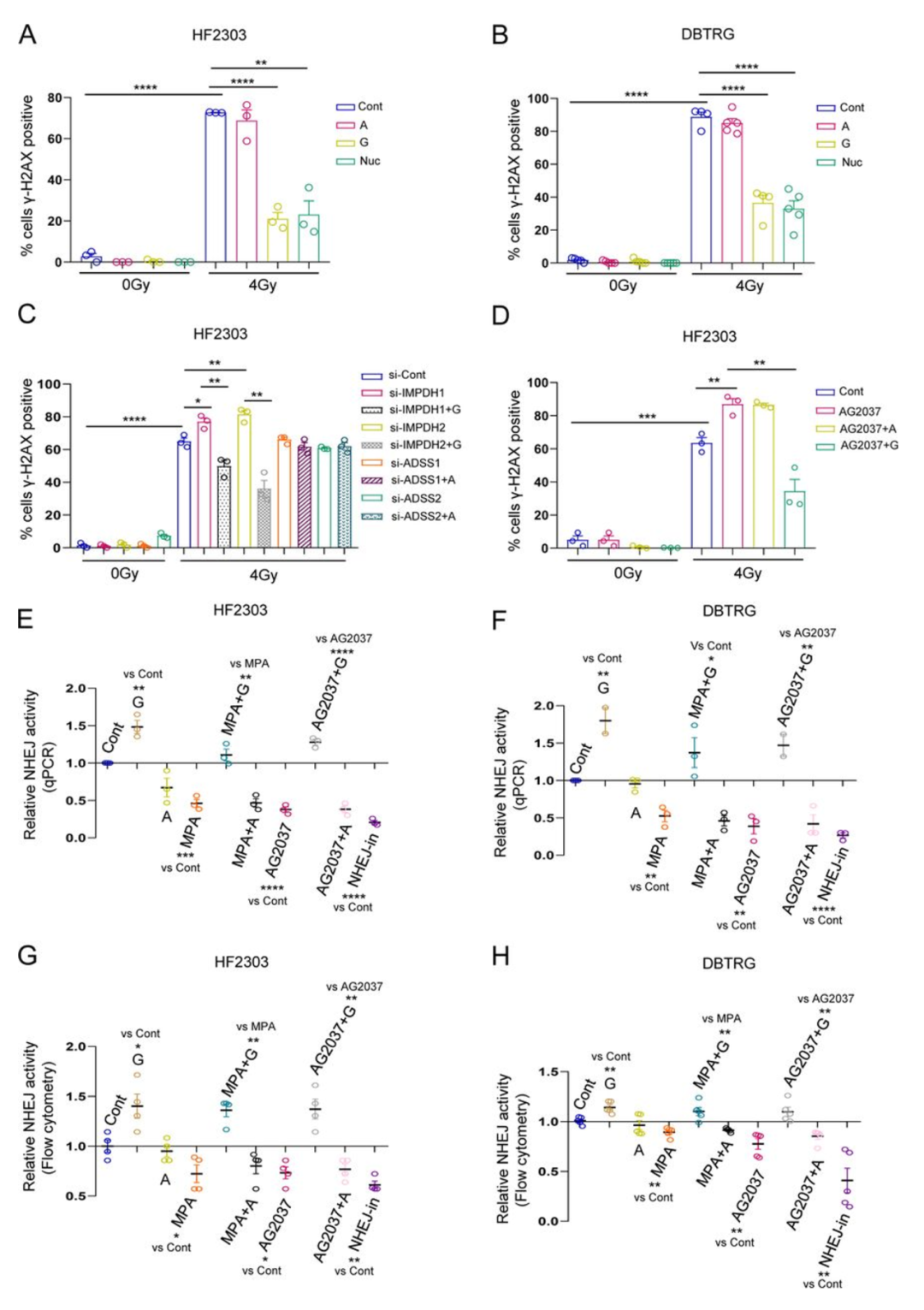
图1
DSBs主要通过NHEJ非同源末端连接或HR同源重组途径修复。在多个细胞系中通过qPCR和流式细胞分析得知,GTP的补充强化了NHEJ(而ATP无效)。两种分析方法发现,不论是单独抑制GTP合成的MPA还是对GTP/ATP合成共同抑制的AG2037造成的GTP减低都会降低NHEJ修复效率。GTP(而非ATP)的补充解救了两种抑制剂的效应(图1E-H)。调整GTP水平并不影响HR的效率(图S1H)或者RT诱导的RAD51灶(HR标志物)形成(图S1I-J)。由此,作者得出结论,是GTP(而非ATP)通过增强NHEJ来促进DSBs修复。
结果二:Abi-1-S323的去磷酸化是GTP诱导的NHEJ的关键
在NHEJ的促进上,GTP是核苷酸中独一无二的,据此作者提出假说,GTP在DNA修复中不是作为单纯的核酸前体物质起作用。已知不同于其他核苷酸,GTP可以通过GTP结合蛋白(G蛋白)或者转化为cGMP来激活一些特定通路。为了确认GTP相关信号通路是否将嘌呤水平和NHEJ相联系起来,作者利用患者来源的GBM神经球进行磷酸化蛋白质组学检测来识别由DNA损伤(RT)、GTP补充(鸟苷)诱导的磷酸化事件以及由MPA对GTP阻断诱导的去磷酸化事件(图2A)。作者识别出约17000个磷酸化位点,其中2182个为RT后的磷酸化,1913个为RT后的去磷酸化。其中1个RT诱导磷酸化的位点被GTP补充所增强,而被MPA介导的GTP减少所阻断;而其中5个RT诱导去磷酸化位点被GTP补充所增加,而被MPA介导的GTP减少所阻断(图2A)。在这六个备选点位中,作者优先选择了Ablinteractor1(Abi-1,图2B)上的Serine323位点,Abi-1先前从未报道与DNA损伤修复相关,但明确可与小G蛋白Rac1结合。再加上Rac1可对生理浓度的GTP水平变化产生反应。与预想相符,作者磷酸化蛋白质组学数据显示,Abi-1-S323的去磷酸化被MPA介导GTP减少所阻断,而被鸟苷带来的GTP增加所回调(图2B)。至此,作者提出假说,Abi-1-S323的去磷酸化是GTP调节DNA损伤修复信号通路中的节点事件。
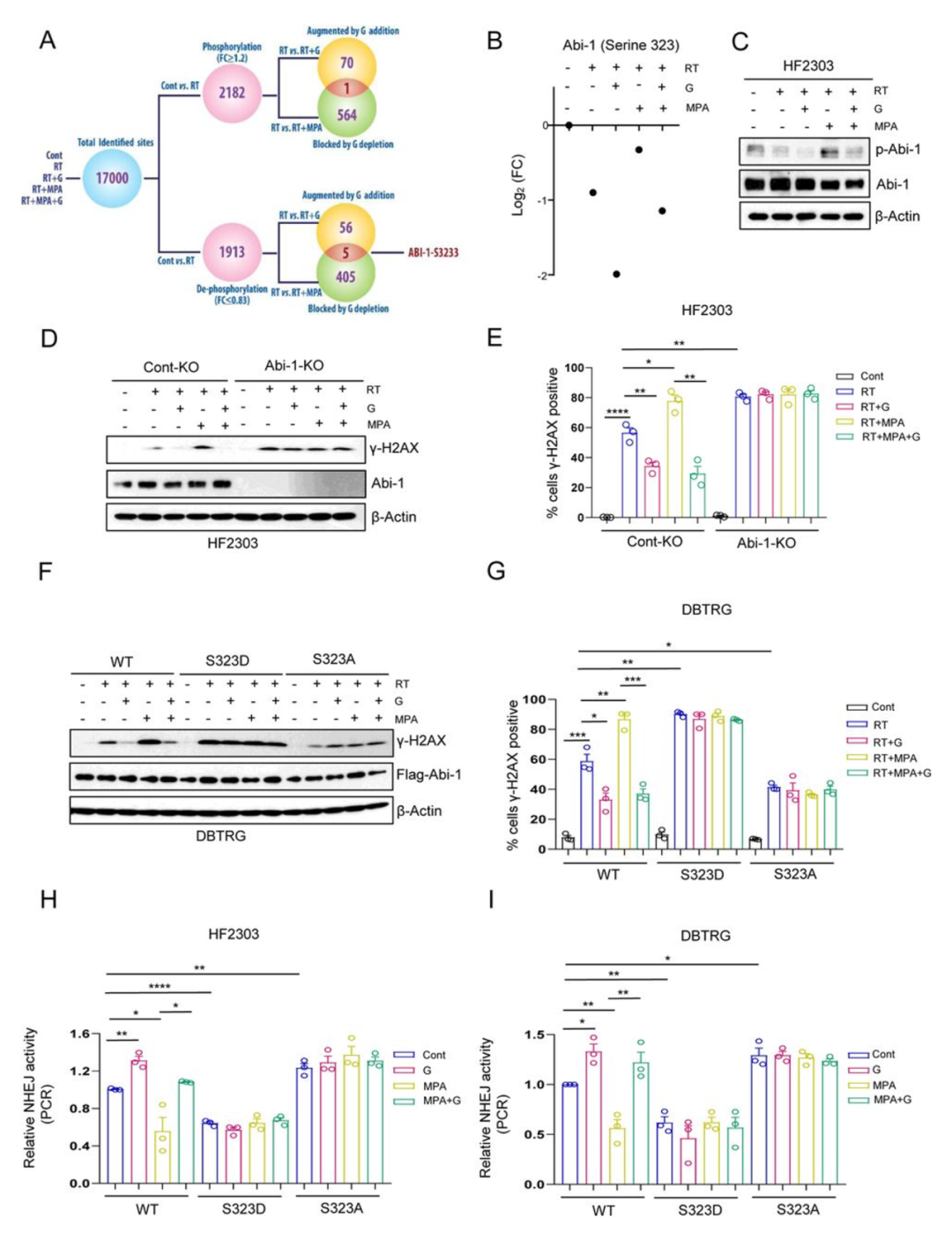
图2
为了查明其中机制,作者构建了Abi-1-S323蛋白质磷酸化(p-Abi-1)抗体,并通过WesternBlot验证了其特异性。在野生型Abi-1或磷酸化模拟Abi-1的模型中,这种抗体在Abi-1分子量的位置显示出了期望条带,而在去磷酸化模拟Abi-1模型或Abi-1敲除模型中没有显示(图S2A)。p-Abi-1在RT后减少,并可在鸟苷补充后进一步减少,但在GTP减少时有所回调(图2C,图S2B),作者利用p-Abi-1-S323抗体对该现象的验证进一步确认了磷酸化蛋白质组学数据。
为了研究Abi-1及其去磷酸化在DDR中的作用,作者利用CRISPR-CAS9构建了Abi-1敲除(KO)的GBM细胞系和神经球(图2D)。通过免疫印迹检测γ-H2AX蛋白水平以及利用IF检测γ-H2AX灶来评估DSBs修复情况。与之前的研究和初步发现(图1)一致,对照-KO GBM细胞中RT诱导DSBs的修复被GTP补充所促进,但被GTP耗竭所阻断。Abi-1-KO GBM细胞在RT后γ-H2AX升高,提示DSB修复速度减慢。此外,RT后Abi-1-KO GBM细胞中γ-H2AX的量不再受GTP补充或耗竭的影响(图2D E,图S2C D)。这些结果表明,Abi-1在GTP介导的DNA损伤修复中发挥作用。为具体探究Abi-1-S323去磷酸化,作者在Abi-1-KO GBM细胞中重新表达WT、S323D/磷酸化拟态、及S323A/去磷酸化拟态的Abi-1,并通过免疫印迹和病灶形成评估RT后的γ-H2AX水平。重新表达磷酸化拟态Abi-1(S323D)减缓了DSB修复,而去磷酸化拟态Abi-1(S323A)则增强了DSB修复。GTP补充或耗竭仅影响表达了Abi-1-WT的细胞的DSB修复,但不影响表达任何点突变Abi-1的细胞的DSB修复(图2F G,图S2E F)。
为了研究Abi-1去磷酸化调控哪种DSB途径,作者采用了线性化质粒末端连接试验来检测NHEJ,并采用RAD51病灶试验来检测HR。表达Abi-1-S323A的细胞增强了NHEJ,而表达Abi-S323D的细胞削弱了NHEJ。此外,在表达这些点突变体的细胞中,NHEJ不再受到GTP补充或耗竭的影响(图2H I)。突变的Abi-1-S323不影响HR活性(图S2G H)。这些发现共同表明,GTP诱导的Abi-1-S323去磷酸化对于通过NHEJ进行DSB修复至关重要。此外,由于GTP调节不再影响表达点突变Abi-1细胞中的DSB修复或NHEJ,表明GTP在促进NHEJ途径DSB修复中的主要作用是通过促进Abi-1去磷酸化,而不是作为新合成的核酸的前体物质。
结果三:GTP依赖性Rac1调控Abi-1-S323去磷酸化
接下来,作者试图找出GTP水平的变化是如何导致Abi-1-S323的去磷酸化以促进DNA修复的。Abi-1通常与G蛋白Rac1结合,并促进肌动蛋白细胞骨架重塑和细胞迁移。为了确定Rac1是否能促进Abi-1-S323去磷酸化和随后的DNA修复,作者引入了3个Rac1质粒(Rac1-WT、显性负性突变体Rac1-T17N和活性突变体Rac1-Q61L),并使用Rac1活性测定确认它们的活性(图S3A)。GTP前体鸟苷的补充会增加辐射后的Rac1活性,但在表达Rac1-WT质粒的细胞中,MPA对GTP的耗竭会阻断Rac1活性。表达组成型活性Rac1-Q61L突变体的细胞保持较高的Rac1活性,而表达显性负性突变体Rac1-T17N的细胞在放疗后显示出最小的Rac1活性。在表达Rac1-Q61L或Rac1-T17N的细胞中,GTP的补充或耗竭都不会改变Rac1的活性(图3A, 图S3B)。此外,与表达Rac1-WT的细胞相比,表达显性负性Rac1-T17N的细胞RT后γ-H2AX水平更高,而表达Rac1-Q61L的细胞RT后γ-H2AX水平更低(图3B,图S3C)。在表达组成型活性或显性负性Rac1的细胞中,补充或消耗GTP水平不再影响RT后γ-H2AX水平。这些数据表明,依赖于GTP的Rac1活性对RT诱导的DSB修复非常重要。
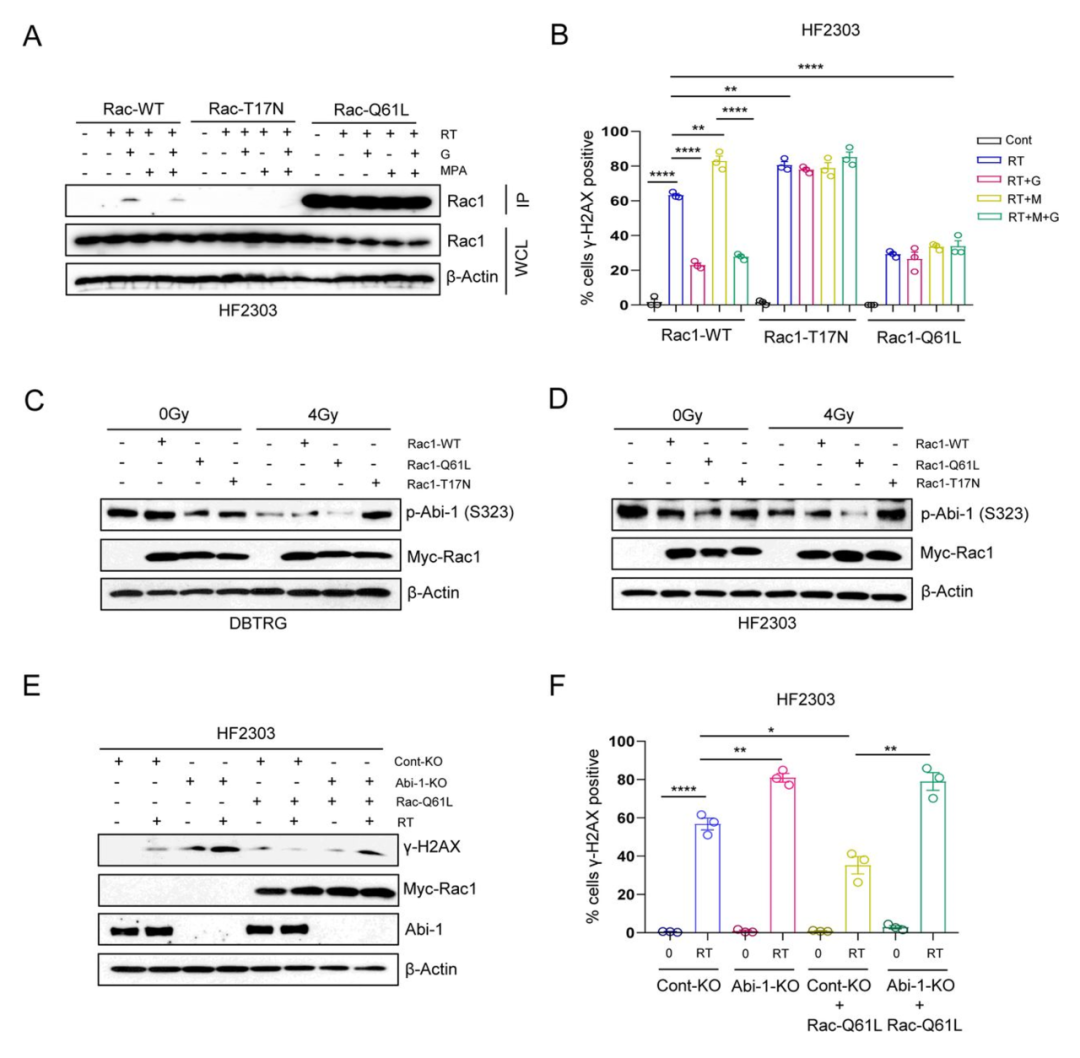
图3
为了探究Abi-1-S323的去磷酸化是否发生在Rac1激活的下游,作者进行了表观实验。辐照表达Rac1-WT的细胞导致Abi-1-S323的去磷酸化。这种去磷酸化作用在表达组成型活性Rac1-Q61L的细胞中增强,但在表达显性负性Rac1-T17N的细胞中被阻断。此外,根据γ-H2AX蛋白水平(图3E,图S3D)和病灶形成水平(图3F,图S3E)评估,在缺乏Abi-1的细胞中表达组成型活性Rac1-Q61L不再促进RT后的DSB修复。由此,作者的数据证实了GTP激活的Rac1促进Abi-1-S323的去磷酸化,进而促进RT诱导的DSB修复。
结果四:蛋白磷酸酶5介导GTP/Rac1下游Abi-1(S323)的去磷酸化
接下来,作者试图确定是哪种磷酸酶负责介导Rac1依赖性的Abi-1-S323去磷酸化。作者用抑制蛋白磷酸酶1/2A/4/5的okadaic酸(OA)以及抑制蛋白磷酸酶2A/ 4的fostriecin(Fos)处理GBM细胞(DBTRG)或神经球(HF2303)。作者发现okadaic酸能阻断RT诱导的Abi-1-S323的去磷酸,但fostriecin不能(图4A,图S4A)。这一发现表明PP1或PP5可能是Rac1依赖性Abi-1-S323去磷酸化的原因。为了进一步阐明负责的磷酸酶,作者单独沉默PP1、PP2A、PP4和PP5,发现只有沉默PP5才能阻断RT诱导的p-Abi-1-S323的减少(图4B,图S4B)。这些数据表明PP5负责DNA损伤后的Abi-1-S323去磷酸化,这与之前Rac1可结合并激活PP5的报道一致。
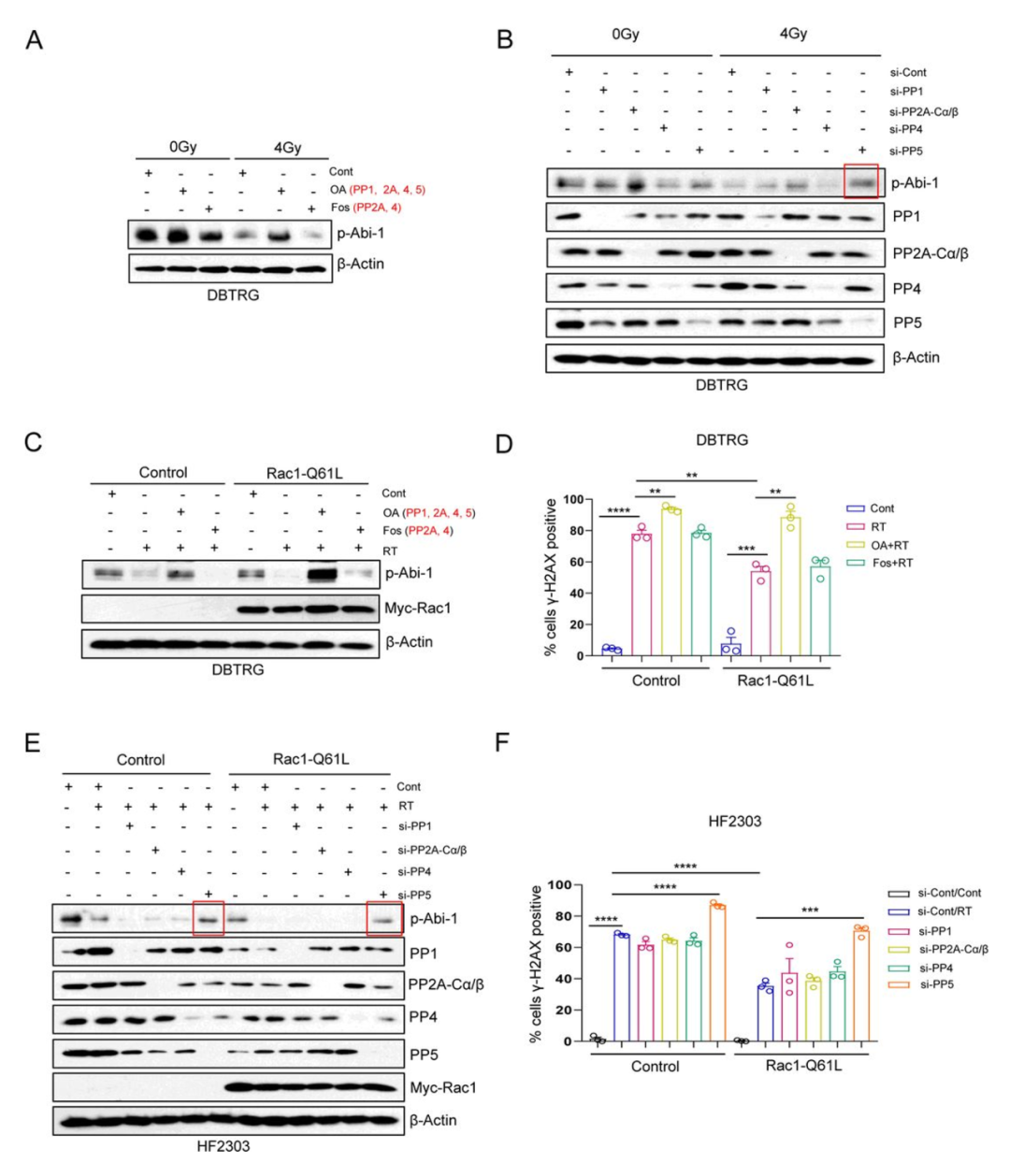
图4
接下来,作者确定了PP5诱导的Abi-1-S323去磷酸化是否依赖于GTP-Rac1激活并负责DSB修复。组成型活性Rac1-Q61L的表达增强了Abi-1-S323的去磷酸化,并增强了RT后的DSB修复。这些作用被okadaic酸阻断,但不被fostriecin阻断(图4C D,图S4C D)。同样作者证实,只有沉默PP5才能阻断表达组成型活性Rac1-Q61L细胞中增强的DSB修复(图4E F,图S4E F)。作者的数据表明存在一个信号转导轴,其中高GTP水平促进Rac1活性,从而导致PP5介导的Abi-1-S323去磷酸化,进而通过NHEJ激活DSB修复。
结果五:Rac1活性影响GBM治疗反应
Rac1的活性增加与许多癌症的治疗抵抗性和生存期缩短有关,这表明它可能是癌症治疗的一个有希望的靶点。为了研究GBM中的这些关联,作者证实了Rac1及其下游靶标的高转录表达与GBM患者较低的生存率相关(图S5A B)。作者查询了DepMap,发现在超过1000种癌细胞系中,Rac1与Abi-1是共同必需的(图S5C),表明Rac1的某些致癌特性可能是由于它调节Abi-1的能力。为了进一步证明Rac1和p-Abi-1-S323在GBM治疗抵抗中的作用,作者求助于梅奥诊所脑肿瘤患者衍生异种移植(PDX)国家资源,利用由此数据库中GBM PDX构建的组织微阵列,作者查询了Abi-1-S323和另两种Rac1下游蛋白PAK1和PAK2的磷酸化水平,这两种蛋白在被Rac1激活时都会被磷酸化。作者在侧腹和颅内Abi-1基因敲除GBM肿瘤中测试了新的p-Abi-S323抗体,证实它适用于免疫组化IHC,并证实这两种模型中都没有信号(图S5D)。在GBM PDX组织样本中,作者发现PAK1和PAK2磷酸化的增加与替莫唑胺(TMZ)以及TMZ和RT联合治疗的耐药性相关。相比之下,磷酸化Abi-1-S323的缺乏与TMZ和TMZ/RT治疗耐药性相关(图5A-F)。
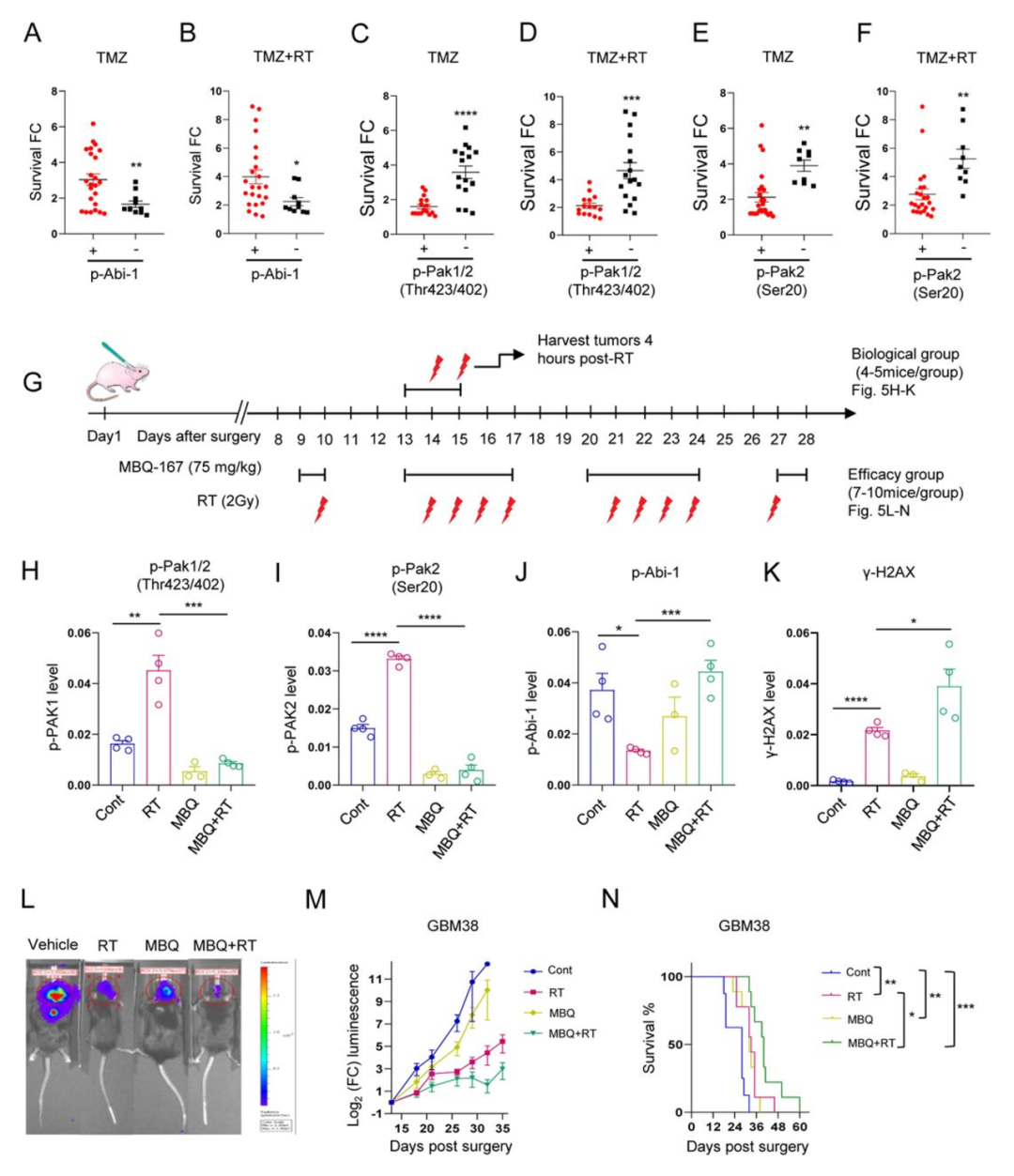
图5
接下来,作者试图干预这一途径,以减缓GBM DNA修复并克服治疗耐药性。作者构建了高RT抗性的原位GBM PDX(GBM38)并将其分为四组:单独RT;单用Rac1抑制剂MBQ-167(MBQ);RT和MBQ-167联合治疗;溶剂对照。为了评估参与其中的靶点、信号传导和DNA修复,对小鼠采用每日两次RT(2Gy/次)和每日3次MBQ-167(75mg/kg/天)的简易方案,并在最后一次RT结束 4小时后收集肿瘤样本(图5G)。RT增加了PAK1和PAK2的磷酸化,降低Abi-1-S323的磷酸化(图5H J,图S5E),MBQ-167对Rac1的抑制阻止了RT诱导的PAK1和PAK2磷酸化以及p-Abi-1-S323的去磷酸化。γ-H2AX染色的增加证明MBQ-167治疗还减缓了RT诱导的DSB的修复(图5K,图S5E),这表明Rac1活性增加可促进RT后Abi-1-S323的去磷酸化和DSB修复。
为了评估GBM肿瘤生长的减缓,作者对荷瘤小鼠进行10次分割RT和/或12剂MBQ-167的处理(图5G)。单独RT减缓了肿瘤生长和延长了小鼠的存活期,当RT与Rac1联合使用时,抑制剂效应增强(图5L-N)。在原位GBM模型(HF2303,图S5F-I)中也观察到了类似结果。因此,激活的Rac1通过Abi-1-S323去磷酸化来促进DNA修复和RT抵抗,抑制Rac1可以阻断这种信号传导并提高疗效。
结果六:Abi-1影响GBM的基因毒性疗效
发现Abi-1调节DNA修复并且Rac1活性影响GBM治疗抵抗性后,作者着手确定Abi-1及其去磷酸化是否调节GBMs对基因毒性治疗的反应。对DepMap中胶质瘤细胞系的分析表明,Abi-1表达与抗癌药物博来霉素耐药性相关,博来霉素通过诱导DSB介导细胞死亡(图S6A)。此外,高Abi-1表达与GBM PDXs中对TMZ和TMZ+RT的耐受性相关(图S6B C),这表明Abi-1可能直接保护GBM免受基因毒性效应。
然后利用Abi-1敲除KO细胞系和神经球,作者开始探究Abi-1如何调节GBM对多种基因毒性疗法的反应。在HF2303神经球(图6A)和永生化DBTRG细胞(图S6D)中,Abi-1的敲除增强多种剂量的RT敏感性。此外,Abi-1敲除增强了博来霉素(图6B,图S6E)、TMZ(图6C,图S6F)和TMZ+RT(4Gy)(图6D,图S6G)的作用,降低了基因毒性疗法在两种细胞模型中的IC50。为确保Abi-1敲除选择性地使GBM对基因毒性治疗敏感,而不是更普遍的细胞死亡,作者用非基因毒性的化学疗法重复了这些实验。Abi-1敲除不影响细胞对紫杉醇或长春新碱的敏感性(图S6H-K),这两种药物是通过靶向微管表现出抗癌能力的。为明确是Abi-1的去磷酸化介导了对基因毒性治疗的耐药性,作者将Abi-1-WT、Abi-1-S323D和Abi-1-S323A重新转入Abi-1-KO细胞中。与Abi-1-WT相比,去磷酸化拟态Abi-1-S323A的再表达恢复了对基因毒性疗法的抗性,而磷酸化拟态Abi-1-S323D增强了其基因毒性敏感性(图6E-H,图S6L-O),这表明Abi-1-S323的去磷酸化有助于介导GBM对基因毒性疗法的抵抗。
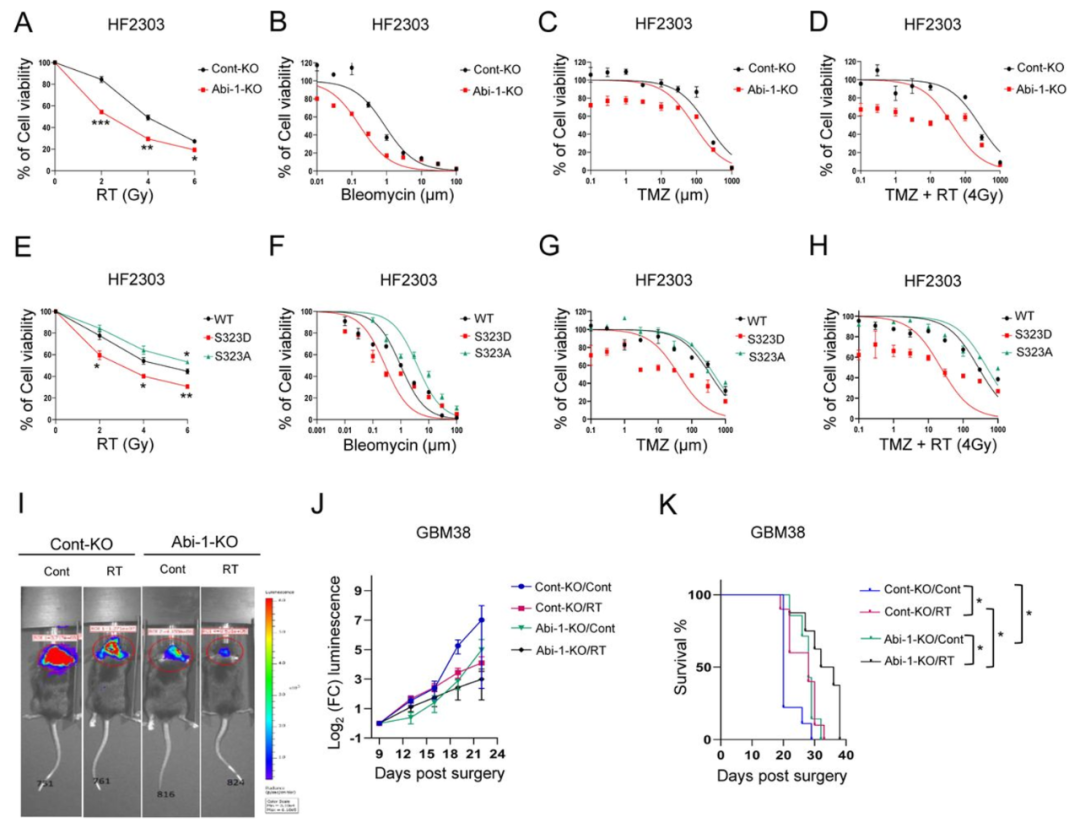
图6
为了在体内研究这种生物学特性,作者使用对照组及Abi-1敲除的GBM38细胞生成原位GBM肿瘤,并在肿瘤形成后给予小鼠6次放射治疗(图6I J)。在对照组肿瘤中,与对照治疗相比,RT减缓了肿瘤生长并延长了小鼠生存期(P=0.01;Cont-KO/对照与Con-KO/RT;图6K)。与对照组肿瘤相比,Abi-1敲除可显著减缓肿瘤生长并延长小鼠存活期(P=0.02;Cont-KO/对照与Abi-1-KO/对照;图6K),放疗后可进一步提高存活率(P=0.03;Abi-1-KO/对照与Abi-1-KO/RT)。据此得知,Abi-1在体内介导GBM对基因毒性治疗的抗性。
结果七:GTP通过Rac1/Abi-1保护正常组织免受基因毒性
在GBM等癌症中,DDR会减轻遗传毒性疗法的疗效。然而在非转化的组织中,DDR能保护细胞免受DNA复制、细胞代谢和暴露于环境因素所遭受的基因毒性应激,以及基因毒性抗癌疗法对正常组织的脱靶效应。为了确定GTP/Rac1/Abi-1信号轴是否介导正常组织中的DDR,作者首先在GTP补充和不补充情况下照射小鼠肠道。RT诱导了肠道中显著的γ-H2AX阳性灶,补充GTP可显著减少γ-H2AX灶(P<0.0001,图7A,图S7A)。与作者在GBM中的发现一致,RT也诱导了肠道组织中GTP依赖性的Abi-1去磷酸化(图7B)。作者在正常人星形胶质细胞中发现了类似的RT诱导和GTP依赖性Abi-1的去磷酸化(图7C)。因为星形胶质细胞与肠胶质细胞相比更适合转染,作者选择该模型来确认Abi-1的去磷酸化依赖于正常组织中的Rac1活性。组成型活性Rac1-Q61L的表达促进正常人星形胶质细胞中RT后Abi-1-S323的去磷酸化,显性负性Rac1-T17N的表达阻断了Abi-1-S323去磷酸化(图7D),这与在GBMs中的发现一致(图3)。这些数据表明GTP通过Rac1/Abi-1通路促进DNA修复,并保护正常组织免受基因毒性效应。
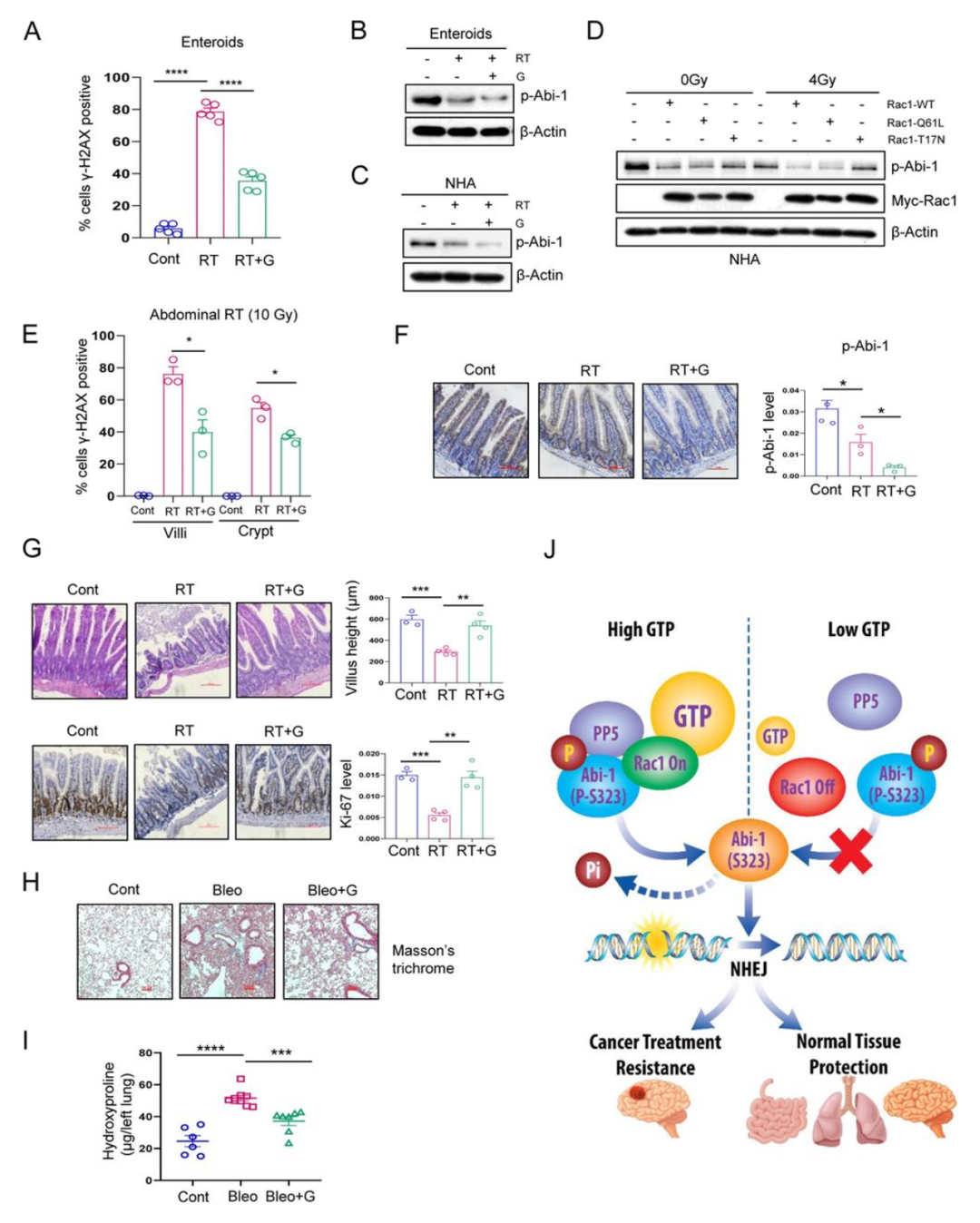
图7
作者使用了两种小鼠体内模型来验证体外发现。作者研究了RT诱导的胃肠道损伤和博来霉素诱导的肺纤维化,这是临床常见的基因毒性治疗的副作用。对C57BL/J6小鼠进行了全身和腹部照射,并在放疗4小时后摘取空肠进行IF或IHC染色。腹部(图7E,图S7B)和全身辐射(图S7C D)都引起肠绒毛和隐窝上显著的DSB,而通过给予鸟苷来增加GTP水平可以挽救这种情况。同样,腹部放疗引起Abi-1-S323去磷酸化,而施用鸟苷增强了这种去磷酸化(图7F)。为了进一步研究放疗引起的肠损伤,作者在腹部放疗后第14天处死小鼠并摘取空肠,这是放疗诱导明显肠道损伤的时间点。RT引起绒毛缩短(图7G;上图),并通过ki-67染色测量发现隐窝增殖减少(图7G;下图),两者都可以通过补充鸟苷逆转。作者选择博来霉素诱导的肺纤维化作为基因毒性疗法诱导正常组织损伤的正交模型(图S7F)。不出所料,通过Masson三色染色(图7H)和羟脯氨酸测定法评估(图7I),博来霉素处理的小鼠出现了肺纤维化。用鸟苷处理补充GTP池显著降低了博来霉素诱导的纤维化程度。因此,作者证明了GTP/Rac1/Abi-1通路可以保护正常组织免受基因毒性应激。
研究结论
细胞内高水平GTP通过激活G蛋白Rac1-PP5-Abi-1信号通路提高细胞DNA损伤NHEJ修复效率。
GTP以外源鸟苷形式的补充可以在恶性/非恶性细胞/组织中提供辐射抗性。
本研究第一次揭示了核苷酸与DNA损伤修复相关的部分分子机制,为新的辅助疗法/联合疗法的临床应用提供依据。
原文出处:Zhou W, Zhao Z, Lin A, Yang JZ, Xu J, Wilder-Romans K, Yang A, Li J, Solanki S, Speth JM, Walker N, Scott AJ, Wang L, Wen B, Andren A, Zhang L, Kothari AU, Yao Y, Peterson ER, Korimerla N, Werner CK, Ullrich A, Liang J, Jacobson J, Palavalasa S, O'Brien AM, Elaimy AL, Ferris SP, Zhao SG, Sarkaria JN, Gyorffy B, Zhang S, Al-Holou WN, Umemura Y, Morgan MA, Lawrence TS, Lyssiotis CA, Peters-Golden M, Shah YM, Wahl DR. GTP signaling links metabolism, DNA repair, and responses to genotoxic stress. Cancer Discov. 2023 Oct 30. doi: 10.1158/2159-8290.CD-23-0437. Epub ahead of print. PMID: 37902550.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#DNA修复# #GTP# #基因毒性疗法#
73