Nature:中国学者领衔,人类“泛基因组”成果登上封面!
2023-05-14 BioMed科技 BioMed科技 发表于上海

由国际人类泛基因组参考联盟牵头,由美国国立卫生研究院国家人类基因组研究所 (NHGRI) 资助的研究人员发布了一份人类“泛基因组”草稿——这是一个新的、可用的基因组学参考,它结合了来自不同祖先背景的
人类基因组计划于2003年完成了第一个完整的人类基因组序列,在具有里程碑意义的人类基因组计划的第一份基因组草图发布20多年后,由国际人类泛基因组参考联盟牵头,由美国国立卫生研究院国家人类基因组研究所 (NHGRI) 资助的研究人员发布了一份人类“泛基因组”草稿——这是一个新的、可用的基因组学参考,它结合了来自不同祖先背景的 47 个人的遗传物质,以便更深入、更准确地理解全球基因组多样性。值得一提的是,该篇Nature封面总共有119位学者,第一作者为中国学者Wen-Wei Liao,通讯作者分别为Erik Garrison, Tobias Marschall, Ira M. Hall, Heng Li, Benedict Paten。

Figure 1. Nature封面
什么是泛基因组?
它是一组来自许多个体的基因组,放在一起以显示序列相同或不同的位置。人类泛基因组草案由47个基因组组成,该项目将持续到 2024 年,届时研究人员计划发布包含 350 个人基因组信息的最终泛基因组。目前,由于依赖于单一参考基因组,一些变异对研究人员来说基本上是不可见的。
等等,什么是参考基因组?
它是一种地图。当研究人员对某人的DNA进行测序时,他们会根据它们在参考基因组中的适合位置得到很多片段。这有点像通过查看解剖学教科书来查看每块骨头适合的位置来组装骨架。对于绝大多数骨骼来说,这很好,但有些人有额外的骨骼,例如教科书中没有的颈肋骨。目前,当科学家绘制来自患者的序列图时,总是有一小部分序列,有时是相当大的一部分,无法被绘制出来。
参考基因组基于谁的DNA?
参考基因组本应由20名匿名捐赠者的DNA混合而成,但最终,其中73%来自一个人。后来的分析表明,那个人是非裔美国人,而且第二大捐助者(大约6%)主要是东亚血统。
科学家已经对数百万个基因组进行了测序,为什么还没有泛基因组?
这是因为测序的许多基因组远未完成——事实上,当人类基因组计划宣布“完成”时,单一参考基因组仅完成了92%。当时只能对短的DNA片段进行测序,而且由于大部分基因组是高度重复的,因此许多这些小片段无法重新组装。泛基因组项目使用了产生更长片段的方法,称为“读取”。因此,泛基因组基于99%完整的极高质量序列。
我们为什么需要它?——【了解基因组变异】
每个人的基因组略有不同——与下一个人相比平均相差约 0.4%——了解这些差异可以深入了解他们的健康状况,有助于诊断疾病、预测医疗结果和指导治疗。使用泛基因组参考将提高科学家在未来研究中检测和理解变异的能力。
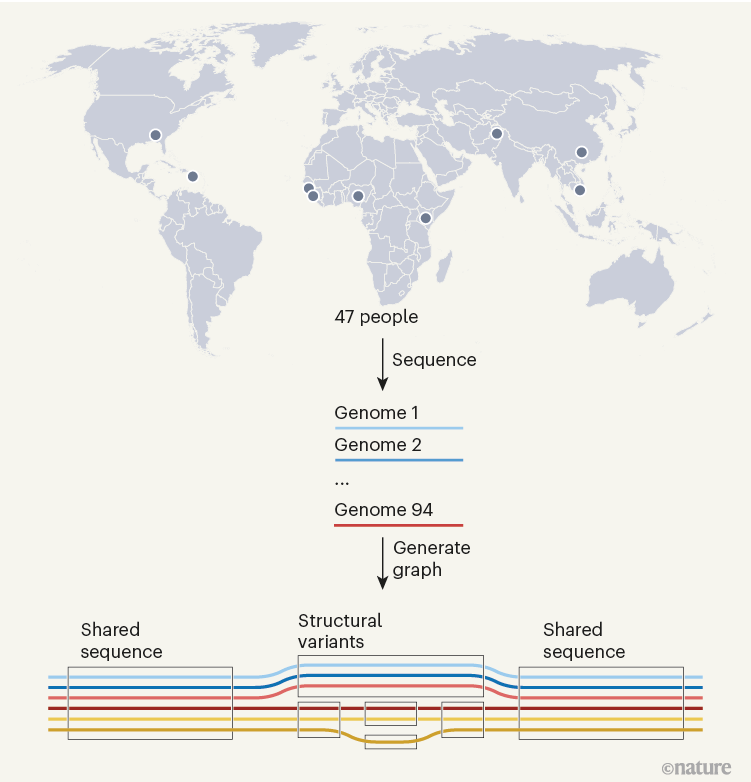
Figure 2. 人类泛基因组
通常,当科学家和临床医生研究个体的基因组以寻找变异时,他们会将个体的 DNA 与标准参考的 DNA 进行比较,以确定一个或多个碱基对的差异所在。到目前为止,参考基因组主要由每个人类染色体的单个序列表示,主要来自一个个体。但是,这个参考已有将近 20 年的历史,并且从根本上受到限制,因为它不能代表人类群体中存在的丰富的遗传变异。这在基因组分析中引入了一个称为参考偏差的问题。
相比之下,新的泛基因组是一个参考,它结合了来自不同祖先背景的 47 个个体的基因组。泛基因组在序列具有相同碱基的区域看起来像线性参考,并扩展以显示存在差异的区域。它同时代表了人类基因组序列的许多不同版本,并为科学家提供了一个更准确的比较点,用于比较某些人群中存在的变异,而不是其他人群中存在的变异。
在泛基因组参考中添加的 1.19 亿个新碱基中,大约有 9000 万个来自结构变异。结构变异很复杂,可能是序列倒置、插入、缺失或串联重复——两个或多个碱基重复多次的片段。这些新碱基将帮助研究人员研究基因组中以前没有参考的区域,并有可能在未来的研究中将结构变异与疾病联系起来。
与使用标准参考的检测相比,使用泛基因组参考进行基因组分析可将结构变异的检测提高 104%。由于泛基因组中存在的数据量增加,泛基因组参考还提高了调用小变体(那些只有几个碱基长)的准确性约 34%。
每个人都携带一对染色体——一组遗传自母亲,一组遗传自父亲。泛基因组参考中的个体基因组包含单倍型解析信息,这意味着它可以自信地区分两组父母的染色体——这是一项重大的科学壮举。掌握这些信息将有助于科学家更好地了解各种基因和疾病是如何遗传的。
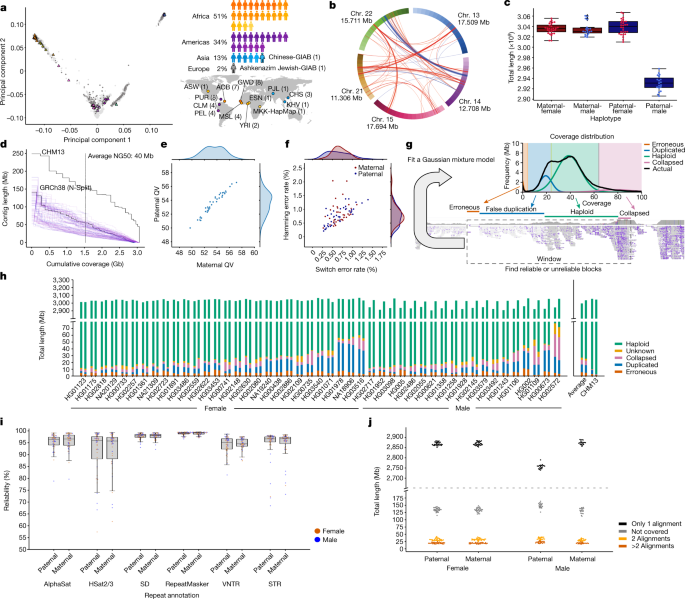
图 1. 呈现 47 个准确且接近完整的多样化二倍体人类基因组组合
【创建泛基因组】
通过开发先进的计算技术将多个基因组序列对齐到一个称为泛基因组图的结构中的可用参考,使泛基因组成为可能。Paten 和 UCSC 计算基因组学实验室的研究人员帮助领导 HPRC 努力开发创建这种泛基因组图结构所需的算法方法。由于该项目使用的方法,泛基因组参考中的所有基因组都具有极高的质量和准确性,覆盖了每个人类基因组的 99% 以上,准确率超过 99%。通过 Asri 的管道后,各种基因组通过复杂的算法方法编译成泛基因组图结构。在视觉上,图形基因组允许研究人员将各种参考序列中的差异视为其他共享路径中的发散区域。

图 2. 组件的转录组注释
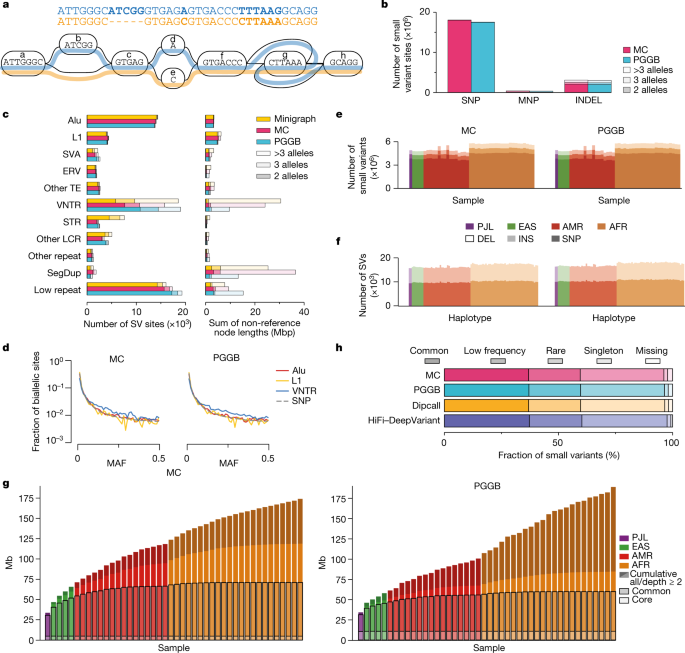
图 3. 泛基因组图代表不同的变异

图 4. 泛基因组图评估
建立可访问的资源
泛基因组草案中的所有前 47 个二倍体基因组都来自参与千人基因组计划 (1000G) 的个人,这是一项有影响力的工作,根据公开同意的样本创建了一个常见的人类遗传变异目录,并于 2015 年完成。这些样本的同意状态允许任何研究人员访问资源,而无需通常伴随基因组研究的隐私障碍,目的是让尽可能多的人可以访问泛基因组。
除了关注可访问性外,HPRC 项目还有一个专门的道德团队,专注于该项目的社会和法律影响。他们正在努力预测具有挑战性的问题并帮助指导知情同意,优先研究不同样本,探索与临床采用有关的可能监管问题,并与国际和土著社区合作,将他们的基因组序列纳入这些更广泛的努力。
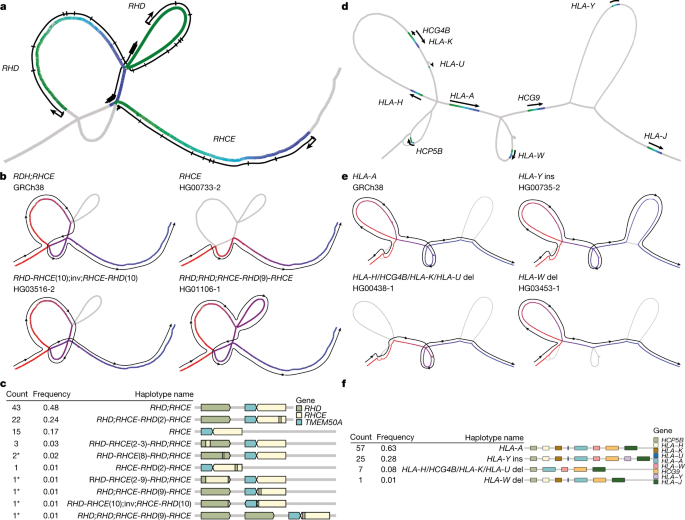
图 5. 可视化复杂的泛基因组位点

图 6. 泛基因组辅助分析短读 WGS 数据的性能提升
继续遗产和未来的工作
人类泛基因组是加州大学圣克鲁兹分校的科学家为了解构成人类生命基础的生物密码而进行的数十年努力的延续。研究人员正在朝着到 2024 年完成完整泛基因组的目标取得进展。该团队正在招募新个体来代表一些未包括在千人基因组计划中的人群,尤其是中东和非洲血统的人群。
除了完成最终的泛基因组参考,研究人员还在努力组建一个国际人类泛基因组项目,该项目将与世界各地的研究人员建立合作伙伴关系。这些伙伴关系将包括双向技能和知识交流,旨在将创建高质量参考基因组所需的技能和技术交到全球研究人员手中,以便他们能够开展自己的研究。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




