

Leber氏先天性黑蒙症(LCA)是一组遗传性视网膜变性疾病,是儿童遗传性失明的最常见原因。LCA一般出现在儿童时期,并导致严重的视力丧失和潜在的失明。近年来,发现数种与LCA相关的致病基因,主要包括GUCY2D、RPE65和CRX等至少18种不同突变基因。
图片来源:iScience
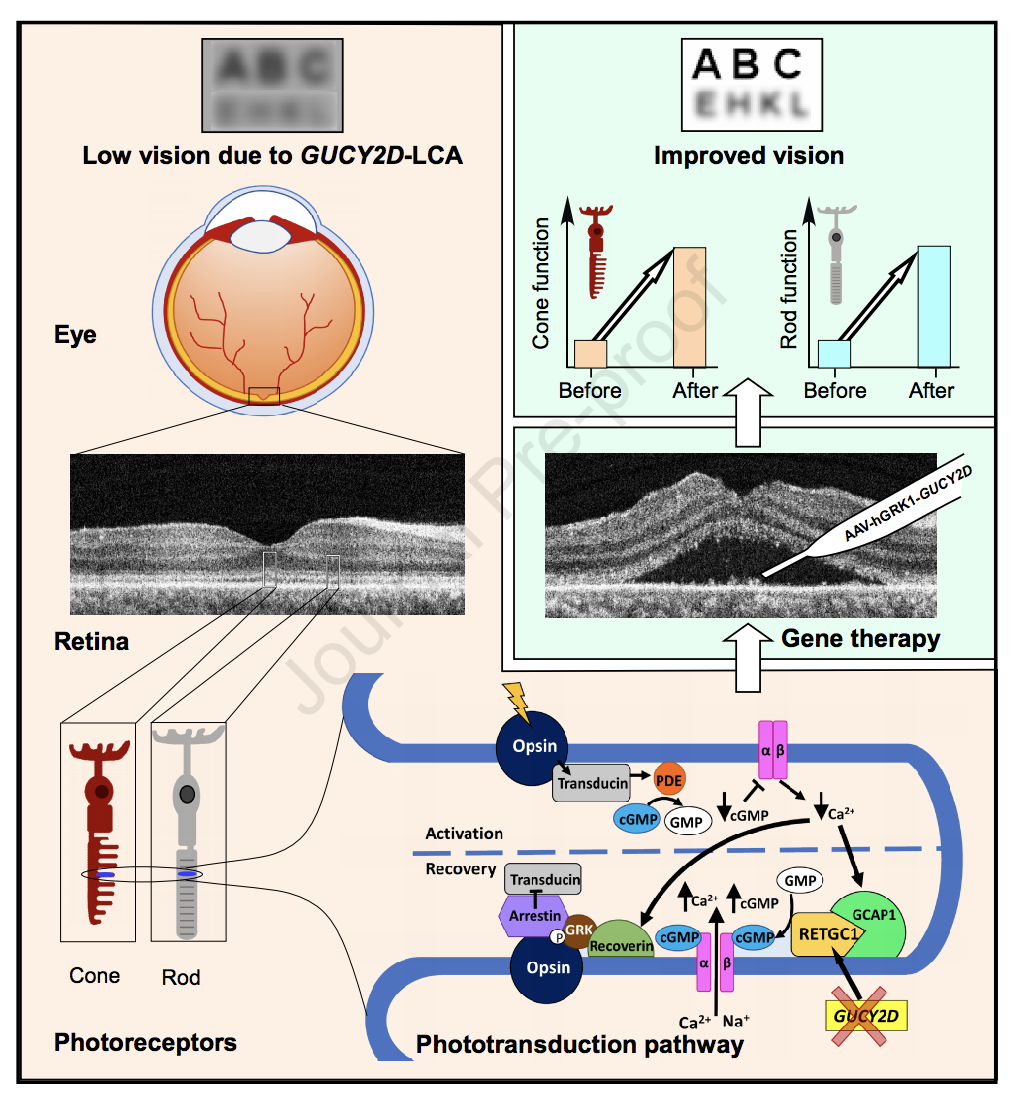
近日,发表在iScience杂志的一项研究显示,目前正在进行的由GUCY2D基因突变引起的LCA的基因疗法完成了人体临床试验。接受该基因治疗的患者视力均得到改善,而没有严重的副作用。

GUCY2D基因正常拷贝编码了一种酶,视网膜中的光敏杆状细胞和视锥细胞使用该酶将光转换为电化学信号。尽管患者在严重视力丧失的情况下生活了十几年,但许多光敏性视网膜细胞仍然可以存活并完好无损。因此,通过基因疗法添加功能性复制的GUCY2D可以使这些细胞再次恢复其功能。
研究人员将携带人GUCY2D基因的重组腺相关病毒血清型5(rAAV5)载体,通过视网膜下注射的方式送入3名视力严重下降、眼球震颤但视网膜结构保留的成年患者眼睛中,并在术后9个月对安全性和疗效进行监测。
随访2年后,研究人员描述了3名患者9个月的发现。第一位患者的杆状细胞的光敏性显着提高,杆状细胞的光敏性比视锥细胞高,并且主要是造成弱光或“夜视”。同时,该患者出现瞳孔对光的敏感性有所改善。第二名患者在基因治疗后约两个月开始显示杆状细胞的光敏感性,虽然较小但持续增加。第三位患者的杆状细胞敏感性没有改善,但在九个月的随访期内视力明显改善。

未治疗和治疗的患者眼睛的视力.
研究人员表示,GUCY2D基因疗法首次试验的这些初步结果令人鼓舞。这3名患者使用的基因治疗剂量是研究人员计划在研究中使用的最低剂量,未来可能使用更高剂量进行治疗,希望能看到更好的安全性和疗效。
当然,早在2017年,美国食品和药物管理局(FDA)专家小组一致投票表示,遗传性盲症的基因治疗效果超过其风险。之后,全球首个上市的眼科基因疗法Luxturna,也用于治疗遗传性失明。Luxturna利用腺相关病毒(AAV)技术将工作拷贝的RPE65基因直接引入患者的视网膜细胞中,产生正常的RPE65酶,从而使具有足够数量的活视网膜细胞患者恢复和改善视力。
此外,2020年3月,基因疗法AGN-151587(EDIT-101)用于遗传性失明疾病Leber氏先天性黑蒙症10型(LCA10)的1/2期临床已完成首例患者给药。这是世界上第一个在人体内给药的CRISPR基因组编辑药物。据悉,迄今为止,该基因疗法只应用在一例患者体内,结果仍然未知。如果耐受良好且成功,则患者可能有资格接受另一只眼睛的治疗。
原始出处
Samuel G. Jacobson et al. Safety and Improved Efficacy Signals following Gene Therapy in Childhood Blindness Caused by GUCY2D Mutations, iScience (2021). DOI: 10.1016/j.isci.2021.102409
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#SCIE#
79
#ISC#
94