

内容摘要
神经元核内包涵体病
-
神经元核内包涵体病(neuronal intranuclear inclusion disease,NIID)又被称为神经元核内透明包涵体病(neuronal intranuclear hyaline inclusion disease,NIHID),或者是核内包涵体病(intranuclear inclusion body disease,INIBD);
-
目前NIID的病因和发病机制尚不明确;
-
是一种罕见的缓慢进展性神经系统变性疾病,其特征为广泛存在于CNS和周围神经系统以及其他器官组织细胞核内的嗜酸性透明包涵体;
-
通常慢性或亚急性起病,男女比例约为1:2;
-
MRI、皮肤活检以及基因检测可协助诊断。
临床表现
Sone等分析57例成人NIID的临床和病理特点,根据遗传学特点将其分为散发型和家族型。散发性NIID患者发病年龄51~76岁,以痴呆症状最为突出(94.7%),其次为瞳孔缩小(94.4%)、共济失调(52.8%)和意识障碍(39.5%)。家族性NIID患者中,发病年龄小于40岁者,以四肢无力最常见(100%),被称为肢体无力;在发病年龄超过40岁,以痴呆症状最为突出(100%),被称为痴呆组。Tian等根据临床表现将家族型NIID又增加新分组-帕金森组。
Takahashi-Fujigasaki等将NIID根据患者发病年龄分为三型:
-
婴儿型,在婴儿期发病,临床病程相对较短(<5岁发病,病程不超过10年);
-
青少年型,在婴儿期或青少年期发病,临床病程长达10年或10年以上;
-
成人型,>50岁以后发病。我国以成人型NIID为主。
成人型NIID临床症状复杂多变,具有异质性,呈慢性或亚急性过程,表现为中枢神经系统以及自主神经受累等。其中发作性意识障碍具有重要提示意义。
影像学表现
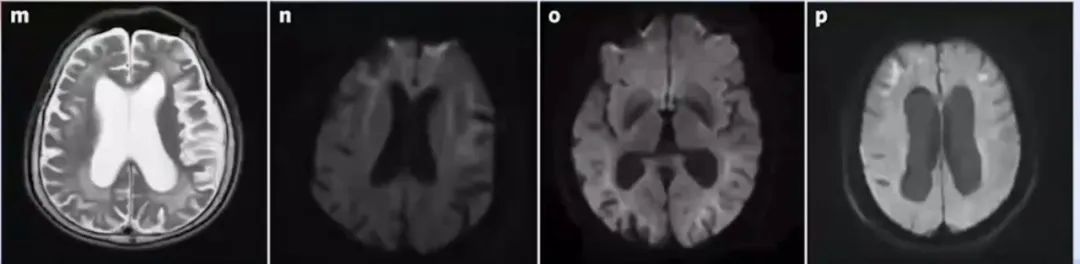
成人型NIID的特征性影像学表现为DWI序列上位于大脑皮交界区的曲线样高信号,由额叶向后延伸,即“飘带征”;
尽管“飘带征”为NIID的一个特征性影像学表现,但可能只出现在疾病晚期或某些特定时期。有学者提出以额叶病变为主的白质脑病可能是NIID早期更为敏感的诊断指标;
此外,脑萎缩也是NIID常见影像学改变之一,尤其在儿童或青少年起病的患者中可能是唯一改变。
确诊手段
以往诊断NIID通常依靠尸检、脑活检或直肠活检及腓肠神经活检,费用高,且常发生复杂的穿透性损伤。
2011年首次通过皮肤活检发现患者的成纤维细胞、汗腺细胞和脂肪细胞核内有嗜酸性包涵体存在,从而使NIID的生前诊断率明显提高。
NIID分子机制
-
自1984年NIID概念的首次提出至2018年一直尚未发现此病的致病基因。
-
NOTCH2NLC基因于2018年被首次发现,但其具体功能及致病性尚未清楚,直至发现其与NIID疾病密切相关,其致病性才被认识。
-
NOTCH2NLC基因是位于1q21.1的三个人类特异性NOTCH2相关基因(NOTCH2NLA、NOTCH2NLB和NOTCH2NLC)之一,其在各种胶质细胞中高表达,被认为与人类大脑皮层的进化扩展有关。
-
2019年,Sone等在某家族性NIID中发现NOTCH2NLC基因5’区的GGC异常重复扩增,随后在另外8个家族性NIID及40个散发性NIID中发现相似的扩增。
-
2019年,Tian等通过长读长测序(long read sequencing,LRS)揭示了NIID致病机制与NOTCH2NLC基因中GGC异常重复扩增相关。
-
NIID临床表现的多样性可能与NOTCH2NLC基因5’区域GGC病理性重复次数相关,一般认为GGC重复扩增次数超过60次具有致病性。以震颤为主要临床表现的患者GGC重复次数接近60次;以帕金森综合征为主要临床表现患者其重复次数约在80次左右;以认知功能障碍为主要表现者其重复次数在120次;肢体无力型患者GGC重复次数在200次。
-
NOTCH2NLC基因与其他神经退行性疾病,如阿尔茨海默病、额颞叶痴呆、帕金森病、帕金森综合征、成人脑白质病变、特发性震颤及多系统萎缩均有关,并在这些患者的病理活检中亦发现了核内嗜酸性包涵体。尽管这对NIID的诊断造成了一定困扰,但对于具有影像学经典“绸带征”的患者,联合病理检查发现存在经典的P62和Ubiquitin染色阳性的嗜酸性核内包涵体以及NOTCH2NLC基因检测,诊断并不困难。
鉴别
脆性X相关震颤/共济失调综合征(fragile X-associated tremor/ataxia syndrome,FXTAS)
FXTAS的致病基因为FMR1,表现为CGG序列异常重复扩增,临床上可显示与NIID相似的症状以及核内包涵体,且有研究者发现FXTAS甚至也能出现大脑皮髓交界区的DWI高信号,目前,尚未有关于FXTAS患者的皮肤组织学检查的研究报道。因此皮肤活检与基因检测对诊断NIID至关重要。
Sone等报道FXTAS中的神经元丢失仅限于浦肯野细胞,并且少突胶质细胞中未观察到核内包涵体,而NIID中的神经元丢失广泛分布于中枢神经系统和周围神经系统,少突胶质细胞中存在p62阳性的核内包涵体。
治疗
NIID尚无有效治疗方法,主要为对症支持治疗;
有文献报道在表现为脑病的病例中短期使用大剂量糖皮质激素冲击可能减轻脑水肿、改善意识状态,但应用效果并不确切。
总结
-
NIID临床症状复杂多样,可单独或同时累及CNS、周围及自主神经系统,最常见痴呆、肢体无力、排尿障碍、发作性意识障碍、共济失调等。
-
影像学皮髓交界区的DWI曲线样高信号具有特征性,对于疑诊患者需建议行皮肤活检和基因检测。
-
皮肤活检是NIID的一种有效的、微创的诊断工具,临床建议取外踝10cm处、3mm厚皮肤活检进行病理检查,取至真皮层及皮下脂肪组织,以便观察到汗腺细胞、纤维母细胞及脂肪细胞中的核内包涵体。光镜下核内嗜酸性透明包涵体,呈圆形,直径为1.5~10μm,位于核仁附近。
-
总之:NIID确断需结合特征性的影像学改变,皮肤活检行HE染色及p62、泛素等免疫组化染色,以及电镜检查,必要时可行FMR1基因及NOTCH2NLC基因检测。
-
治疗上:尽早对症状进行干预治疗可改善生活质量。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



