
肝豆状核变性:症状与体征、病因、流行病学、诊断与治疗
2022-08-18 MedSci原创 MedSci原创

肝豆状核变性(hepatolenticular degeneration,HLD)由Wilson在1912年首先描述,故又称为威尔逊病(Wilson病,Wilson Disease,WD)。是一种常染
肝豆状核变性(hepatolenticular degeneration,HLD)由Wilson在1912年首先描述,故又称为威尔逊病(Wilson病,Wilson Disease,WD)。是一种常染色体隐性遗传的铜代谢障碍性疾病,以铜代谢障碍引起的肝硬化、基底节损害为主的脑变性疾病为特点,对肝豆状核变性发病机制的认识已深入到分子水平。WD的世界范围发病率为1/30 000~1/100 000,致病基因携带者约为1/90。本病在中国较多见。WD好发于青少年,男性比女性稍多,如不恰当治疗将会致残甚至死亡。WD也是少数几种可治的神经遗传病之一,关键是早发现、早诊断、早治疗。 2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,肝豆状核变性被收录其中。
一、一般概述
肝豆状核变性是一种罕见的遗传疾病,其特征是在各种身体组织中储存过量的铜,特别是肝脏、大脑和眼睛的角膜。 这种疾病是进行性的,如果不及时治疗,可能会导致肝脏疾病、中枢神经系统功能障碍和死亡。 早期诊断和治疗可以预防严重的长期残疾和危及生命的并发症。 治疗的目的是减少体内积聚的铜量,并在此后保持正常的铜水平。
二、症状与体征
1、肝功能障碍:
威尔逊病是一种罕见的遗传性疾病,以肝功能障碍开始,其损害从 6 岁开始,但通常在青少年或 20 岁出头时出现临床症状。相关肝病的常见体征包括皮肤、粘膜和眼内膜(巩膜)的黄色变色(黄疸),由于液体潴留异常导致的腿部和腹部肿胀(水肿)(腹水),存在食管中可能出血的异常血管(食管静脉曲张)、瘀伤和长时间出血的趋势以及过度疲劳(疲劳)。一些患有威尔逊病的人可能只有肝功能检查异常,并且可能直到多年后才出现其他症状。
少数受影响的个体可能会出现严重的肝功能衰竭。这种情况在青春期威尔逊氏病患者中最常见,在女性中更常见。这些人可能会迅速出现肝病的体征和症状,通常与红细胞分解(溶血)和精神错乱引起的贫血有关。在这些年轻患者中,眼角膜中特征性的锈褐色沉积物(Kayser-Fleischer 环)可能尚不存在。
在一些患者中,肝病不会自行显现,并且患者会出现神经(脑相关)症状。威尔逊病的常见神经系统症状可能会随着时间的推移而出现和进展,包括震颤、不自主运动、吞咽困难(吞咽困难)、说话困难和发音困难(构音障碍)、缺乏协调、痉挛、肌张力障碍姿势和肌肉僵硬。几乎所有患有威尔逊病神经系统症状的受影响个体的眼睛中都有 Kayser-Fleischer 环,可以由眼科医生识别。
2、神经和精神症状
神经症状以锥体外系损害为突出表现,以舞蹈样动作、手足徐动和肌张力障碍为主,并有面部怪容、张口流涎、吞咽困难、构音障碍、运动迟缓、震颤、肌强直等。震颤可以表现为静止或姿势性的,但不像帕金森病的震颤那样缓慢而有节律性。疾病进展还可有广泛的神经系统损害,出现小脑性共济失调、病理征、腱反射亢进、假性球麻痹、癫痫发作,以及大脑皮质、下丘脑损害体征。精神症状表现为注意力和记忆力减退、智能障碍、反应迟钝、情绪不稳,常伴有强笑、傻笑,也可伴有冲动行为或人格改变。
3、角膜K-F环
角膜色素环是本病的重要体征,出现率达95%以上。K-F环位于巩膜与角膜交界处,呈绿褐色或暗棕色,宽约1.3mm,是铜在后弹力膜沉积而成。
4、其它症状
在年轻女性中,在疾病得到治疗之前,月经可能不会开始或停止。这是由于威尔逊氏病引起的肝病引起的激素代谢普遍紊乱。月经不调、月经不调(闭经)、流产和不孕也很常见。
威尔逊病的其他体征和症状可能包括肾结石和肾小管损伤、过早关节炎,以及其他关节和骨骼受累,包括骨骼变薄(骨质疏松症)和大关节处出现骨质增生(骨赘)。脊柱和四肢关节空间也可能减少。
三、病因
威尔逊病是常染色体隐性遗传。遗传疾病由两种基因决定,一种来自父亲,一种来自母亲。
隐性遗传疾病发生在个体遗传了同一性状的异常基因的两个拷贝时,每个拷贝来自父母一方。如果一个人继承了一个正常基因和一个疾病基因,这个人将成为该疾病的携带者,但通常不会出现症状。每次怀孕,两个携带者父母都通过改变的基因并有受影响的孩子的风险是 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。孩子从父母双方那里获得正常基因的机会是 25%。男性和女性的风险相同。
近亲(近亲)的父母比无关父母携带相同异常基因的可能性更高,这增加了患隐性遗传疾病的孩子的风险。
肝豆状核变性为常染色体隐性遗传性疾病。绝大多数限于同胞一代发病或隔代遗传,罕见连续两代发病。致病基因ATP7B定位于染色体13q14.3,编码一种1411个氨基酸组成的铜转运P型ATP酶。ATP7B基因突变导致ATP酶功能减弱或消失,引致血清铜蓝蛋白(ceruloplasmin,CP)合成减少以及胆道排铜障碍,蓄积在体内的铜离子在肝、脑、肾、角膜等处沉积,引起进行性加重的肝硬化、锥体外系症状、精神症状、肾损害及角膜色素环(Kayser-Fleischerring,K.F环)等。ATP7B基因的变异位点繁多,人类基因组是数据库中记载达300多个位点突变形式。基因突变位点具有种族特异性,因此基因检测位点的选择要有针对性。我国WD患者的ATP7B基因有3个突变热点,即R778L,P992L和T935M,占所有突变的60%左右。近年来有研究发现除ATP7B以外其他基因如COMMD1,XIAP,Atox1等也与该病相关。
疾病状态时,血清中过多的游离铜大量沉积于肝脏内,造成小叶性肝硬化。当肝细胞溶酶体无法容纳时,铜即通过血液向各个器官散布和沉积。基底节的神经元和其正常酶的转运对无机铜的毒性特别敏感,大脑皮质和小脑齿状核对铜的沉积也产生症状。铜对肾脏近端小管的损害可引起氨基酸、蛋白以及钙和磷酸盐的丢失。铜在眼角膜弹力层的沉积产生K-F环。与此同时,肝硬化可产生门静脉高压的一系列变化。
四、流行病学
威尔逊病是一种罕见的疾病,影响同等数量的男性和女性。 这种疾病存在于所有种族和民族中。 尽管估计有所不同,但据信威尔逊氏病发生在全世界大约 30,000 至 40,000 人中。 大约九分之一的人可能是该疾病基因的携带者。 尽管在美国仅诊断出约 2,000-3,000 例病例,但其他受影响的个体可能被误诊为其他神经、肝脏或精神疾病。 根据一项估计,美国实际上可能有 9,000 人受到威尔逊氏病的影响。
五、鉴别诊断
以下疾病的症状可能与威尔逊病的症状相似。比较可能有助于鉴别诊断:
如果患者出现轻度肝病,最常见的误诊是病毒性肝炎。病毒抗原和铜的研究应该区分。如果肝硬化已经确定并且患者饮酒,则通常会错误地诊断为酒精性肝硬化。铜研究应该区分。如果患者出现震颤,可能会被错误地诊断为特发性震颤或早期帕金森病。再次铜研究应该区分。如果出现精神症状,可能会做出药物滥用的错误诊断。同样,铜研究应该有所区别。
有时被误认为是威尔逊氏症的其他疾病如下:
Sydenham 舞蹈病是一种急性的、通常是自限性的疾病,发生在大约 5% 到 10% 的风湿热病例之后。这种疾病通常始于身体一侧或两侧的不平稳、无法控制、不重复的肌肉运动。患者会出现快速、不自主的运动,这些运动会影响步行、手臂运动和说话的方式或风格。笨拙和做鬼脸很常见。
原发性胆汁性胆管炎是一种慢性进行性肝脏疾病,被认为与免疫系统异常有关。这种疾病的最初症状通常包括持续性全身瘙痒、尿色深、大便苍白和黄疸。最终,过量的铜在肝脏中积累,肝脏软组织中出现纤维或颗粒硬化。
重金属中毒通常是由于工业暴露于铜、铝、砷或汞等多种毒素引起的。根据暴露的类型和持续时间,损伤可能发生在肺部、神经系统、皮肤或消化系统。中毒的症状因过度暴露所涉及的金属类型而异。这些包括头痛、恶心、头晕、关节和肌肉疼痛、谵妄、癫痫发作和各种其他症状。
神经棘红细胞增多症是一种非常罕见的神经肌肉和血液系统遗传疾病。产生异常血细胞(棘红细胞增多症),肌肉萎缩(萎缩)。这种疾病的主要症状是不受控制的快速肌肉运动(肌萎缩性舞蹈病)。最初,面部、嘴巴和舌头会出现细微的不自主运动(抽搐)。这些会慢慢发展为躯干和四肢的严重、不受控制的快速运动(舞蹈症)。大约 50% 的 Levine-Critchley 综合征患者有癫痫发作。
亨廷顿舞蹈病(亨廷顿舞蹈病)是一种遗传性、进行性退行性神经系统疾病。最初会有性格变化和不自主的快速肌肉运动。随着时间的推移,言语和记忆力受损,不自主的肌肉运动变得更加频繁和明显。随着亨廷顿病的进展,认知能力和痴呆症进一步丧失。这种疾病的症状通常在成年期开始,通常在四十岁以后。
抽动秽语综合征是一种以重复性运动和发声抽动为特征的神经性运动障碍。最初的症状通常发生在儿童时期,是快速眨眼或做鬼脸。症状还可能包括四肢、肩膀、面部和随意肌的不自主运动。一些患有图雷特综合症的人可能会不由自主地发声;这些可能是口齿不清的声音或单词。抽动秽语综合征不是进行性或退行性疾病;症状往往是多变的,并遵循一个长期的消长过程。发病通常发生在 16 岁之前。
脑性麻痹是一种神经肌肉疾病,是在早期发育或出生时大脑受伤的结果。这种疾病的主要症状是缺乏肌肉控制和协调。脑瘫不是一种进行性疾病。一般来说,婴儿在第一年或第二年可能会出现发育迟缓,并可能出现肌肉无力和肌张力异常。与威尔逊病相关的协调和言语困难可能类似于脑瘫的症状。
六、诊断
威尔逊病可以根据全面的临床评估、完整的患者病史和专门的测试来诊断。此类测试可能包括对眼睛进行裂隙灯检查,以揭示 Kayser-Fleischer 环的存在;对血液(血清)的液体部分进行测试,表明铜蓝蛋白(一种铜蛋白)水平低;以及显示尿液中排出的铜含量异常高的测试。在某些患者中,可能需要进行肝活检以进行铜分析以确认威尔逊病的诊断。使用来自血细胞的 DNA 来寻找差异或相似性模式的分子遗传学研究,称为单倍型分析的程序可以确定受影响患者的全兄弟姐妹是否患有威尔逊病,是否是威尔逊病基因的携带者,或者不是载体。该分析适用于被确定患有威尔逊病的个人的家庭成员。 DNA分析也可用于诊断受影响的患者。在超过一半的患者中,DNA 分析将揭示导致威尔逊病的突变。
尽早诊断威尔逊病很重要。早期诊断和治疗可以避免永久性神经功能障碍和严重肝病。
疾病分类
根据中华医学会神经病学分会帕金森病及运动障碍学组《肝豆状核变性的诊断与治疗指南》,临床分型如下:
(1)肝型:
①持续性血清转氨酶增高;②急性或慢性肝炎;③肝硬化(代偿或失代偿);④暴发性肝功能衰竭(伴或不伴溶血性贫血)。
(2)脑型:
①运动障碍:扭转痉挛、手足徐动、舞蹈症状、步态异常、共济失调等;②口-下颌肌张力障碍:流涎、讲话困难、声音低沉、吞咽障碍等;③精神症状。
(3)其他类型:以肾损害、骨关节肌肉损害或溶血性贫血为主。混合型以上各型的组合。
七、治疗
威尔逊病的治疗是终生的,旨在将铜水平降低到无毒水平,并防止疾病的进展,并试图扭转由于铜在体内积累而出现的任何体征和症状。治疗可分为三部分:一是对有症状的患者进行治疗,二是在受累组织减少铜后维持治疗,三是对无症状患者,从一开始就可以进行维持治疗。
1、饮食治疗
避免进食含铜高的食物如小米、荞麦面、糙米、豆类、坚果类、薯类、菠菜、茄子、南瓜、蕈类、菌藻类、干菜类、干果类、软体动物、贝类、螺类、虾蟹类、动物的肝脏和血、巧克力、可可。某些中药,如龙骨、牡蛎、蜈蚣、全蝎等。
2、药物治疗
威尔逊病的治疗包括三种药物。首先是那些通过尿液排泄从体内去除(螯合)铜的物质,例如青霉胺(Cuprimine)和曲恩汀二盐酸盐(Syprine),其次是锌盐以防止肠道从饮食中吸收铜,第三是四硫代钼酸盐,它们都可以防止吸收铜并结合血液中的有毒铜,使其无毒。
以驱铜药物为主,驱铜及阻止铜吸收的药物主要有两大类药物,一是络合剂,能强力促进体内铜离子排出,如青霉胺、二巯丙磺酸钠、三乙烯-羟化四甲胺、二巯丁二酸等;二是阻止肠道对外源性铜的吸收,如锌剂、四硫钼酸盐。
(1)曲恩汀胶囊主要用于青霉胺不能耐受或用青霉胺复发的肝豆状核变性患者。出现轻至中度肝功能衰竭症状的患者,可采用曲恩汀联合锌治疗 4-6 个月,然后继续单独使用锌或曲恩汀维持治疗。
(2)D-青霉胺(D-penicillamine,PCA):是本病的首选药物,为强效金属螯合剂,在肝脏中可与铜形成无毒复合物,促使其在组织沉积部位被清除,减轻游离状态铜的毒性。青霉胺与组织中的同类子络合成铜-青霉胺复合物,从尿中排出。本药口服易吸收。药物副作用有恶心、过敏反应、重症肌无力、关节病、无疱疮,少数可以引起白细胞减少和再生障碍性贫血。视神经炎、狼疮综合症。剥脱性皮炎、肾病综合征等较严重的毒副作用。另外,当患者首次用药时应做青霉胺皮试,阴性者才能使用。本病需长期甚或终生服药,应注意补充足量维生素B。
(3)二巯基丙磺酸(DMPS):DMPS 5mg/kg溶于5%葡萄糖溶液500ml中缓慢静滴,每日1次,6天为1疗程,2个疗程之间休息1-2天,连续注射6-10个疗程。不良反应主要是食欲减退及轻度恶心、呕吐。可用于有轻、中、重度肝损害和神经精神症状的肝豆状核病患者。
(4)三乙烯-羟化四甲胺(TETA):药理作用与D-青霉胺相似,是用于不能耐受青霉胺治疗时的主要药物。副作用小,但药源困难、价格不菲。
(5)锌制剂(zinc preparations):常用有硫酸锌、醋酸锌、葡萄糖酸锌、甘草锌等。在餐后1 h服药以避免食物影响其吸收,尽量少食粗纤维以及含大量植物酸的食物。锌剂副反应较小,主要有胃肠道刺激、口唇及四肢麻木感、免疫功能降低、血清胆固醇紊乱等。对胎儿无致畸作用。锌剂的缺点是起效慢(4~6个月),严重病例不宜首选。醋酸锌(Galzin)已被批准用于患者的维持治疗。对于没有症状(无症状)的受影响个体或最初使用螯合剂治疗的个体,醋酸锌(Teva Pharmaceuticals 的 Gate 部门生产的 Galzin)用于防止铜从肠道吸收。由于副作用有限,儿童和孕妇通常首选锌疗法。对于一些因胃刺激而不能耐受锌的患者,曲恩汀维持治疗可能更可取。
(6)四硫钼酸盐(tetl.athiomolybdate,TM):能促进体内的金属铜较快排出,改善WD的症状与PCA相当,副作用则比PcA少得多。本药在国外仍未商品化,国内未有使用的经验。主要用于出现神经病的患者最好用四硫代钼酸盐治疗。
(7)中药治疗:大黄、黄连、姜黄、金钱草、泽泻、三七等由于具有利尿及排铜作用而对wD有效,少数患者服药后早期出现腹泻、腹痛。使用中药治疗WD,效果常不满意,中西医结合治疗效果会更好。推荐用于症状前患者、早期或轻症患者、儿童患者以及长期维持治疗。
慢性药物治疗的监测包括随访身体检查、24 小时尿液收集中铜(以及锌治疗者的锌)测量、血液检查以确定未与铜蓝蛋白(游离铜)结合的铜量、定期测量肝功能和血细胞计数。对于那些使用螯合剂的人,还应定期进行尿液分析以寻找尿液中是否存在细胞或蛋白质。通常不需要重复肝活检来跟踪药物治疗的进展。
停药治疗威尔逊病可能会导致铜的快速积聚和危及生命的事件。
3、对症治疗
有震颤和肌强直时可用苯海索口服,对粗大震颤者首选氯硝西泮。肌张力障碍可用苯海索、复方左旋多巴制剂、多巴胺受体激动剂,还可服用氯硝西泮、硝西泮、巴氯芬,局限性肌张力障碍药物治疗。无效者可试用局部注射A型肉毒毒素。有舞蹈样动作和手足徐动症时,可选用氯硝西泮、硝西泮、氟哌啶醇,合用苯海索。对于精神症状明显者可服用抗精神病药奋乃静、利培酮、氟哌啶醇、氯氮平,抑郁患者可用抗抑郁药物。护肝治疗药物也应长期应用。
4、手术治疗
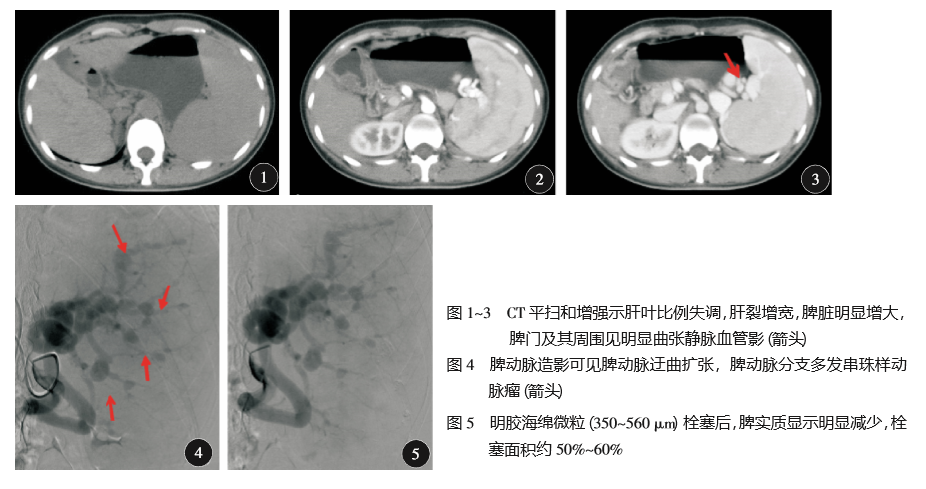
对于有严重脾功能亢进者可行脾切除术,严重肝功能障碍时也可以考虑肝移植治疗。
对于出现严重肝功能衰竭的个体,肝移植可能会挽救生命。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Brewer GJ. Wilson Disease. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:506.
Brewer GJ. Wilson’s Disease: A Clinician’s Guide to Recognition, Diagnosis, and Management. Kluwer Academic Publishing; Boston, 2001.
Brewer GJ. Wilson’s Disease for the Patient and Family: A Patient’s Guide to Wilson’s Disease and Frequently Asked Questions about Copper. Xlibris, Philadelphia; 2001.
Schilsky ML. Wilson’s Disease. In: Diseases of the Liver. Philadelphia, PA: Lippincott-Raven; 1999:1091-1106.
Beers MH, Berkow R., eds. The Merck Manual, 17th ed. Whitehouse Station, NJ: Merck Research Laboratories; 1999:56-8.
Kanski JJ., ed. Clinical Ophthalmology, 4th ed. Woburn, MA: Butterworth-Heinemann; 1999:142.
Fauci AS, et al., eds. Harrison’s Principles of Internal Medicine, 14th Ed. New York, NY: McGraw-Hill, Inc; 1998:2166-69.
Adams, RD, et al., eds. Principles of Neurology. 6th ed. New York, NY: McGraw-Hill, Companies; 1997:969-71.
Bennett JC, Plum F., eds. Cecil Textbook of Medicine. 20th ed. Philadelphia, PA: W.B. Saunders Co; 1996:1131-2.
Schilsky ML. Wilson’s Disease — Genetic Basis of Copper Toxicity and Natural History. In: Seminars in Liver Disease. New York, NY: Thieme Medical Publishers, Inc.: 1996:83-95.
Behrman RE, ed. Nelson Textbook of Pediatrics, 15th ed. Philadelphia, PA: W.B. Saunders Company; 1996:1139-40.
Scriver CR, et al., eds. The Metabolic and Molecular Basis of Inherited Disease. 7th Ed. New York, NY; McGraw-Hill Companies, Inc; 1995:2217-23.
Menkes JH., au., Pine JW, et al., eds. Textbook of Child Neurology, 5th ed. Baltimore, MD: Williams & Wilkins; 1995:118-25.
Leevy CM, et al., eds. Diseases of the Liver and Biliary Tract: Standardization of Nomenclature, Diagnostic Criteria and Prognosis. New York, NY: Raven Press; 1994:112-3.
Abuduxikuer K, Wang JS. Zinc mono-therapy in pre-symptomatic Chinese children with Wilson disease: a single center, retrospective study. PLoS ONE 9(1):2014. e86168. https://doi.org/10.1371/journal.pone.0086168
Brewer, GJ. Treatment of Wilson’s Disease: Our patients deserve better. Expert Opin on Orphan Drugs 2014;2:12.
El-Youssef M, Wilson disease. Mayo Clin Proc. 2003;78:1126-36.
Ferenci P, et al., Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139-42.
Pellecchia MT, et al., Clinical presentation and treatment of Wilson’s disease: a single-centre experience. Eur Neurol. 2003;50:48-52.
Brewer GJ, et al., Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 2003;60:379-85.
Bacon BR, et al., New knowledge of genetic pathogenesis of hemochromatosis and Wilson’s disease. Adv Intern Med. 1999;44:91-116.
Sturniolo GC, et al., Zinc therapy increases duodenal concentrations of metallothionein and iron in Wilson’s disease patients. Am J Gastroenterol. 1999;94:334-38.
Schaefer M, et al., Hepatocyte-specific localization and copper-dependent trafficking of the Wilson’s disease protein in the liver. 1999;276:639-46.
Brewer GJ, et al., Treatment of Wilson’s disease with zinc: XV. Long-term follow-up studies. J Lab Clin Med. 1998;132:264-78.
Brewer GJ, et al., Treatment of Wilson disease with ammonium tetrathiomolybdate. Arch Neurol. 1996;53:1017-25.
Demirkiran M, et al., Neurologic presentation of Wilson disease without Kayser-Fleischer rings. Neurology. 1996;46:1040-3.
Scheinberg IH, et al., Treatment of the neurologic manifestations of Wilson’s disease. Arch Neurol. 1995; 52:339-40.
Schilsky ML, et al., Hepatic transplantation for Wilson’s disease: indication and outcome. Hepatology. 1994;19:583-7.
Yarse JC, et al., Wilson’s disease: current status. Am J Med. 1992;92:643-54.
Brewer GJ, et al., Initial therapy of patients with Wilson’s disease with tetrathiomolybdate. Arch Neurol. 1991;48:42-7.
Tankanow RM, Pathophysiology and treatment of Wilson’s disease. Clin Pharm. 1991;10:839-49.
Schilsky ML, et al., Prognosis of Wilsonian chronic active hepatitis. Gastroenterology. 1991;100:762-7.
Woods SE, Wilson’s disease. Am Family Physician. 1989;40:171-8.
Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Wilson Disease. Entry No: 277900. Last Edited 07/09/2016. Available at: http://omim.org/entry/277900.Accessed March 7, 2018.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#诊断与治疗#
108
#肝豆状核变性##罕见病#
168
#流行病#
99
#变性#
144