121种“常见的”罕见病名录,以及我国主要罕见病介绍,请速收藏并转发
2020-12-02 MedSci原创 MedSci原创

中华医学会医学遗传学分会提出的中国罕见病定义,罕见病即患病率低于1/500000或新生儿发病率低于1/10000的疾病。世界上有超过7000种罕见疾病,而且数量在不断增加,每年大约有250种新疾病加入
中华医学会医学遗传学分会提出的中国罕见病定义,罕见病即患病率低于1/500000或新生儿发病率低于1/10000的疾病。世界上有超过7000种罕见疾病,而且数量在不断增加,每年大约有250种新疾病加入名单中。据估算,全球受罕见病影响的人群有2.63亿—4.46亿人,中国约有2000万人。
相关话题:#罕见病#
因罕见病的发病率低、有关专业知识认知有限,易造成误诊或诊断周期延长。约有80%的罕见病由基因缺陷导致,仅有不到5%的罕见病能有效干预或治疗。
2016年,上海市卫计委组织专家论证,以入选病种基本符合中国罕见病定义、具有明确诊断的技术方法、具有一定疗效的干预措施为主要原则,确定将56种疾病纳入《上海市主要罕见病名录(2016年版)》。
上海市主要罕见病名录(2016年版)
|
序号 |
系统 |
中文疾病名称 |
英文疾病名称 |
诊断方法 |
主要治疗原则 |
|
1 |
内分泌与代谢疾病 |
精氨酸酶缺乏症 |
Arginase deficiency |
生化/基因等 |
药物/饮食干预/移植等 |
|
2 |
肾脏疾病 |
非典型溶血性尿毒症 |
Atypical hemolytic uremic syndrome |
生化/基因等 |
药物等 |
|
3 |
内分泌与代谢疾病 |
β-酮硫解酶缺乏症 |
Beta-ketothiolase deficiency |
生化/基因等 |
饮食干预等 |
|
4 |
内分泌与代谢疾病 |
生物素酶缺乏症 |
Biotinidase deficiency |
生化/基因等 |
药物等 |
|
5 |
免疫疾病 |
Chediak-Higashi 综合征 |
Chediak-Higashi syndrome |
基因等 |
移植等 |
|
6 |
免疫疾病 |
原发性慢性肉芽肿病 |
Chronic primary granulomatous disease |
免疫/基因等 |
药物/移植等 |
|
7 |
内分泌与代谢疾病 |
瓜氨酸血症 |
Citrullinemia |
生化/基因等 |
药物/饮食干预/移植等 |
|
8 |
内分泌与代谢疾病 |
先天性肾上腺皮质增生症 |
Congenital adrenal hyperplasia |
生化/基因等 |
药物等 |
|
9 |
内分泌与代谢疾病 |
先天性肾上腺发育不良 |
Congenital adrenal hypoplasia |
生化/基因等 |
药物等 |
|
10 |
内分泌与代谢疾病 |
先天性高胰岛素性低血糖血症 |
Congenital hperinsulinemic hypoglycemia |
生化/基因等 |
药物/手术等 |
|
11 |
血液疾病 |
先天性纯红细胞再生障碍性贫血 |
Diamond-Blackfan anemia |
骨髓/基因等 |
药物/移植等 |
|
12 |
血液疾病 |
先天性角化不良 |
Dyskeratosis congenita |
基因等 |
移植等 |
|
13 |
内分泌与代谢疾病 |
法布雷病 |
Fabry disease |
生化/基因等 |
药物等 |
|
14 |
血液疾病 |
家族性噬血细胞性淋巴组织细胞增生症 |
Familial hemophagocytic lymphohistiocytosis |
血液/基因等 |
移植等 |
|
15 |
血液疾病 |
范可尼贫血 |
Fanconi anemia |
血液/基因等 |
移植等 |
|
16 |
内分泌与代谢疾病 |
半乳糖血症 |
Galactosemia |
生化/基因等 |
饮食干预等 |
|
17 |
内分泌与代谢疾病 |
戈谢病 |
Gaucher's disease |
生化/基因等 |
药物/移植等 |
|
18 |
内分泌与代谢疾病 |
戊二酸血症 I 型 |
Glutaric acidemia I |
生化/基因等 |
饮食干预等 |
|
19 |
内分泌与代谢疾病 |
糖原累积病 |
Glycogen storage diseases |
生化/基因等 |
药物等 |
|
20 |
血液疾病 |
血友病 |
Hemophilia |
凝血/基因等 |
输血/药物等 |
|
21 |
消化疾病 |
肝豆状核变性 |
Hepatolenticular degeneration |
生化/基因等 |
药物/饮食干预等 |
|
22 |
皮肤疾病 |
遗传性大疱性表皮松解症 |
Hereditary epidermolysis bullosa |
病理等 |
药物等 |
|
23 |
内分泌与代谢疾病 |
全羧化酶合成酶缺乏症 |
Holocarboxylase synthetase deficiency |
生化/基因等 |
药物等 |
|
24 |
骨骼疾病 |
低碱性磷酸脂酶血症 |
Hypophosphatasia |
生化/基因等 |
药物等 |
|
25 |
骨骼疾病 |
低磷性佝偻病 |
Hypophosphatemic rickets |
生化/基因等 |
药物等 |
|
26 |
特发性肺动脉高压症 |
Idiopathic pulmonary arterial hypertension |
影像等 |
药物等 |
|
|
27 |
内分泌与代谢疾病 |
异戊酸血症 |
Isovaleric acidemia |
生化/基因等 |
饮食干预等 |
|
28 |
内分泌与代谢疾病 |
莱伦氏综合征 |
Laron syndrome |
生化/基因等 |
药物等 |
|
29 |
内分泌与代谢疾病 |
长链3-羟酰基辅酶A脱氢酶缺乏症 |
Long chain 3-hydroxyacyl-CoA dehydrogenase deficiency |
生化/基因等 |
饮食干预等 |
|
30 |
内分泌与代谢疾病 |
溶酶体酸性脂肪酶缺乏症 |
Lysosomal acid lipase deficiency |
生化/基因等 |
药物/饮食干预等 |
|
31 |
内分泌与代谢疾病 |
枫糖尿症 |
Maple syrup urine disease |
生化/基因等 |
饮食干预等 |
|
32 |
内分泌与代谢疾病 |
中链酰基辅酶A脱氢酶缺乏症 |
Medium chain acyl-CoA dehydrogenase deficiency |
生化/基因等 |
饮食干预/药物等 |
|
33 |
内分泌与代谢疾病 |
甲基丙二酸血症 |
Methylmalonic acidemia |
生化/基因等 |
饮食干预/药物等 |
|
34 |
内分泌与代谢疾病 |
黏多糖贮积症 |
Mucopolysaccharidoses |
生化/基因等 |
移植/药物等 |
|
35 |
内分泌与代谢疾病 |
多种酰基辅酶A脱氢酶缺乏症 |
Multiple acyl-CoA dehydrogenase deficiency |
生化/基因等 |
药物/饮食干预等 |
|
36 |
内分泌与代谢疾病 |
尼曼匹克病 |
Niemann-Pick disease |
生化/基因等 |
药物/移植等 |
|
37 |
五官疾病 |
非综合征性耳聋 |
Non-syndromic deafness |
基因等 |
手术等 |
|
38 |
内分泌与代谢疾病 |
Noonan综合征 |
Noonan syndrome |
生化/基因等 |
药物等 |
|
39 |
内分泌与代谢疾病 |
鸟氨酸氨甲酰基转移酶缺乏症 |
Ornithine transcarbamylase deficiency |
生化/基因等 |
饮食干预/药物等 |
|
40 |
骨骼疾病 |
成骨不全症 |
Osteogenesis imperfecta |
基因等 |
药物等 |
|
41 |
血液疾病 |
阵发性睡眠性血红蛋白尿 |
Paroxysmal nocturnal hemoglobinuia |
血液免疫等 |
药物等 |
|
42 |
内分泌与代谢疾病 |
苯丙酮尿症 |
Phenylketouria |
生化/基因等 |
饮食干预等 |
|
43 |
内分泌与代谢疾病 |
Prader-Willi综合征 |
Prader-Willi syndrome |
基因等 |
药物等 |
|
44 |
内分泌与代谢疾病 |
原发性肉碱缺乏症 |
Primary carnitine deficiency |
生化/基因等 |
药物/饮食干预等 |
|
45 |
内分泌与代谢疾病 |
丙酸血症 |
Propionic acidemia |
生化/基因等 |
饮食干预/药物等 |
|
46 |
免疫疾病 |
重症联合免疫缺陷 |
Severe combined immunodeficiency |
血液免疫/基因等 |
移植等 |
|
47 |
血液疾病 |
重症先天性粒细胞缺乏症 |
Severe congenital neutropenia |
血液/基因等 |
移植等 |
|
48 |
血液疾病 |
先天性中性粒细胞减少伴胰腺机能不全综合征 |
Shwachman-Diamond syndrome |
血液/基因等 |
移植等 |
|
49 |
内分泌与代谢疾病 |
Silver-Russell 综合征 |
Silver-Russell syndrome |
基因等 |
药物/手术等 |
|
50 |
内分泌与代谢疾病 |
四氢生物蝶呤缺乏症 |
Tetrahydrobiopterin deficiency |
生化/基因等 |
药物等 |
|
51 |
内分泌与代谢疾病 |
酪氨酸血症 |
Tyrosinemia |
生化/基因等 |
饮食干预/移植等 |
|
52 |
内分泌与代谢疾病 |
极长链酰基辅酶A脱氢酶缺乏症 |
Very long chain acyl-CoA dehydrogenase deficiency |
生化/基因等 |
饮食干预/药物等 |
|
53 |
免疫疾病 |
湿疹血小板减少伴免疫缺陷综合征 |
Wiskott-Aldrich syndrome |
基因等 |
移植等 |
|
54 |
免疫疾病 |
X-连锁无丙种球蛋白血症 |
X-linked agammaglobulinemia |
血液免疫/基因等 |
药物等 |
|
55 |
免疫疾病 |
X-连锁高IgM血症 |
X-linked hyper IgM syndrome |
基因等 |
移植等 |
|
56 |
免疫疾病 |
X-连锁淋巴增生症 |
X-linked lymphoproliferative disease |
基因等 |
移植等 |
2018年5月22日,国家卫生健康委员会、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局等五部门联合发布了《第一批罕见病》目录,共涉及121种疾病。全球预计有超过3亿名罕见病患者,中国为1680多万。目前已经明确的罕见病有7000多种,其中80%为遗传病。
第一批罕见病目录
| 序 | 中文名称 | 英文名称 |
| 1 | 21-羟化酶缺乏症 | 21-Hydroxylase Deficiency |
| 2 | 白化病 | Albinism |
| 3 | Alport 综合征 | Alport Syndrome |
| 4 | 肌萎缩侧索硬化 | Amyotrophic Lateral Sclerosis |
| 5 | Angelman 氏症候群(天使综合征) | Angelman Syndrome |
| 6 | 精氨酸酶缺乏症 | Arginase Deficiency |
| 7 | 热纳综合征(窒息性胸腔失养症) | Asphyxiating Thoracic Dystrophy (Jeune Syndrome) |
| 8 | 非典型溶血性尿毒症 | Atypical Hemolytic Uremic Syndrome |
| 9 | 自身免疫性脑炎 | Autoimmune Encephalitis |
| 10 | 自身免疫性垂体炎 | Autoimmune Hypophysitis |
| 11 | 自身免疫性胰岛素受体病 | Autoimmune Insulin Receptopathy (Type B insulin resistance) |
| 12 | β-酮硫解酶缺乏症 | Beta-ketothiolase Deficiency |
| 13 | 生物素酶缺乏症 | Biotinidase Deficiency |
| 14 | 心脏离子通道病 | Cardic Ion Channelopathies |
| 15 | 原发性肉碱缺乏症 | Carnitine Deficiency |
| 16 | Castleman病 | Castleman Disease |
| 17 | 腓骨肌萎缩症 | Charcot-Marie-Tooth Disease |
| 18 | 瓜氨酸血症 | Citrullinemia |
| 19 | 先天性肾上腺发育不良 | Congenital Adrenal Hypoplasia |
| 20 | 先天性高胰岛素性低血糖血症 | Congenital Hyperinsulinemic Hypoglycemia |
| 21 | 先天性肌无力综合征 | Congenital Myasthenic Syndrome |
| 22 | 先天性肌强直(非营养不良性肌强直综合征) | Congenital Myotonia Syndrome (Non-Dystrophic Myotonia, NDM) |
| 23 | 先天性脊柱侧弯 | Congenital Scoliosis |
| 24 | 冠状动脉扩张病 | Coronary Artery Ectasia |
| 25 | 先天性纯红细胞再生障碍性贫血 | Diamond-Blackfan Anemia |
| 26 | Erdheim-Chester病 | Erdheim-Chester Disease |
| 27 | 法布雷病 | Fabry Disease |
| 28 | 家族性地中海热 | Familial Mediterranean Fever |
| 29 | 范可尼贫血 | Fanconi Anemia |
| 30 | 半乳糖血症 | Galactosemia |
| 31 | 戈谢病 | Gaucher’s Disease |
| 32 | 全身型重症肌无力 | Generalized Myasthenia Gravis |
| 33 | Gitelman 综合征 | Gitelman Syndrome |
| 34 | 戊二酸血症I型 | Glutaric Acidemia Type I |
| 35 | 糖原累积病(I型、Ⅱ型) | Glycogen Storage Disease (Type I、II) |
| 36 | 血友病 | Hemophilia |
| 37 | 肝豆状核变性 | Hepatolenticular Degeneration(Wilson Disease) |
| 38 | 遗传性血管性水肿 | Hereditary Angioedema (HAE) |
| 39 | 遗传性大疱性表皮松解症 | Hereditary Epidermolysis Bullosa |
| 40 | 遗传性果糖不耐受症 | Hereditary Fructose Intolerance |
| 41 | 遗传性低镁血症 | Hereditary Hypomagnesemia |
| 42 | 遗传性多发脑梗死性痴呆 | Hereditary Multi-infarct Dementia (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, CADASIL) |
| 43 | 遗传性痉挛性截瘫 | Hereditary Spastic Paraplegia |
| 44 | 全羧化酶合成酶缺乏症 | Holocarboxylase Synthetase Deficiency |
| 45 | 同型半胱氨酸血症 | Homocysteinemia |
| 46 | 纯合子家族性高胆固醇血症 | Homozygous Hypercholesterolemia |
| 47 | 亨廷顿舞蹈病 | Huntington Disease |
| 48 | HHH综合征 | Hyperornithinaemia-Hyperammonaemia-Homocitrullinuria Syndrome |
| 49 | 高苯丙氨酸血症 | Hyperphenylalaninemia |
| 50 | 低碱性磷酸酶血症 | Hypophosphatasia |
| 51 | 低磷性佝偻病 | Hypophosphatemic Rickets |
| 52 | 特发性心肌病 | Idiopathic Cardiomyopathy |
| 53 | 特发性低促性腺激素性性腺功能减退症 | Idiopathic Hypogonadotropic Hypogonadism |
| 54 | 特发性肺动脉高压 | Idiopathic Pulmonary Arterial Hypertension |
| 55 | 特发性肺纤维化 | Idiopathic Pulmonary Fibrosis |
| 56 | IgG4相关性疾病 | IgG4 related Disease |
| 57 | 先天性胆汁酸合成障碍 | Inborn Errors of Bile Acid Synthesis |
| 58 | 异戊酸血症 | Isovaleric Acidemia |
| 59 | 卡尔曼综合征 | Kallmann Syndrome |
| 60 | 朗格汉斯组织细胞增生症 | Langerhans Cell Histiocytosis |
| 61 | 莱伦氏综合征 | Laron Syndrome |
| 62 | Leber遗传性视神经病变 | Leber Hereditary Optic Neuropathy |
| 63 | 长链3-羟酰基辅酶A脱氢酶缺乏症 | Long Chain 3-hydroxyacyl-CoA Dehydrogenase Deficiency |
| 64 | 淋巴管肌瘤病 | Lymphangioleiomyomatosis (LAM) |
| 65 | 赖氨酸尿蛋白不耐受症 | Lysinuric Protein Intolerance |
| 66 | 溶酶体酸性脂肪酶缺乏症 | Lysosomal Acid Lipase Deficiency |
| 67 | 枫糖尿症 | Maple Syrup Urine Disease |
| 68 | 马凡综合征 | Marfan Syndrome |
| 69 | McCune-Albrigh综合征 | McCune-Albright Syndrome |
| 70 | 中链酰基辅酶A脱氢酶缺乏症 | Medium Chain Acyl-CoA Dehydrogenase Deficiency |
| 71 | 甲基丙二酸血症 | Methylmalonic Academia |
| 72 | 线粒体脑肌病 | Mitochodrial Encephalomyopathy |
| 73 | 黏多糖贮积症 | Mucopolysaccharidosis |
| 74 | 多灶性运动神经病 | Multifocal Motor Neuropathy |
| 75 | 多种酰基辅酶A脱氢酶缺乏症 | Multiple Acyl-CoA Dehydrogenase Deficiency |
| 76 | 多发性硬化 | Multiple Sclerosis |
| 77 | 多系统萎缩 | Multiple System Atrophy |
| 78 | 肌强直性营养不良 | Myotonic Dystrophy |
| 79 | N-乙酰谷氨酸合成酶缺乏症 | N-acetylglutamate Synthase Deficiency |
| 80 | 新生儿糖尿病 | Neonatal Diabetes Mellitus |
| 81 | 视神经脊髓炎 | Neuromyelitis Optica |
| 82 | 尼曼匹克病 | Niemann-Pick Disease |
| 83 | 非综合征性耳聋 | Non-Syndromic Deafness |
| 84 | Noonan综合征 | Noonan Syndrome |
| 85 | 鸟氨酸氨甲酰基转移酶缺乏症 | Ornithine Transcarbamylase Deficiency |
| 86 | 成骨不全症(脆骨病) | Osteogenesis Imperfecta (Brittle Bone Disease) |
| 87 | 帕金森病(青年型、早发型) | Parkinson Disease (Young-onset , Early-onset) |
| 88 | 阵发性睡眠性血红蛋白尿 | Paroxysmal Nocturnal Hemoglobinuria |
| 89 | 黑斑息肉综合征 | Peutz-Jeghers Syndrome |
| 90 | 苯丙酮尿症 | Phenylketonuria |
| 91 | POEMS综合征 | POEMS Syndrome |
| 92 | 卟啉病 | Porphyria |
| 93 | Prader-Willi综合征 | Prader-Willi Syndrome |
| 94 | 原发性联合免疫缺陷 | Primary Combined Immune Deficiency |
| 95 | 原发性遗传性肌张力不全 | Primary Hereditary Dystonia |
| 96 | 原发性轻链型淀粉样变 | Primary Light Chain Amyloidosis |
| 97 | 进行性家族性肝内胆汁淤积症 | Progressive Familial Intrahepatic Cholestasis |
| 98 | 进行性肌营养不良 | Progressive Muscular Dystrophy |
| 99 | 丙酸血症 | Propionic Acidemia |
| 100 | 肺泡蛋白沉积症 | Pulmonary Alveolar Proteinosis |
| 101 | 肺囊性纤维化 | Pulmonary Cystic Fibrosis |
| 102 | 视网膜色素变性 | Retinitis Pigmentosa |
| 103 | 视网膜母细胞瘤 | Retinoblastoma |
| 104 | 重症先天性粒细胞缺乏症 | Severe Congenital Neutropenia |
| 105 | 婴儿严重肌阵挛性癫痫(Dravet综合征) | Severe Myoclonic Epilepsy in Infancy (Dravet Syndrome) |
| 106 | 镰刀型细胞贫血病 | Sickle Cell Disease |
| 107 | Silver-Russell综合征 | Silver-Russell Syndrome |
| 108 | 谷固醇血症 | Sitosterolemia |
| 109 | 脊髓延髓肌萎缩症(肯尼迪病) | Spinal and Bulbar Muscular Atrophy (Kennedy Disease) |
| 110 | 脊髓性肌萎缩症 | Spinal Muscular Atrophy |
| 111 | 脊髓小脑性共济失调 | Spinocerebellar Ataxia |
| 112 | 系统性硬化症 | Systemic Sclerosis |
| 113 | 四氢生物蝶呤缺乏症 | Tetrahydrobiopterin Deficiency |
| 114 | 结节性硬化症 | Tuberous Sclerosis Complex |
| 115 | 原发性酪氨酸血症 | Tyrosinemia |
| 116 | 极长链酰基辅酶A脱氢酶缺乏症 | Very Long Chain Acyl-CoA Dehydrogenase Deficiency |
| 117 | 威廉姆斯综合征 | Williams Syndrome |
| 118 | 湿疹血小板减少伴免疫缺陷综合征 | Wiskott-Aldrich Syndrome |
| 119 | X-连锁无丙种球蛋白血症 | X-linked Agammaglobulinemia |
| 120 | X-连锁肾上腺脑白质营养不良 | X-linked Adrenoleukodystrophy |
| 121 | X-连锁淋巴增生症 | X-linked Lymphoproliferative Disease |
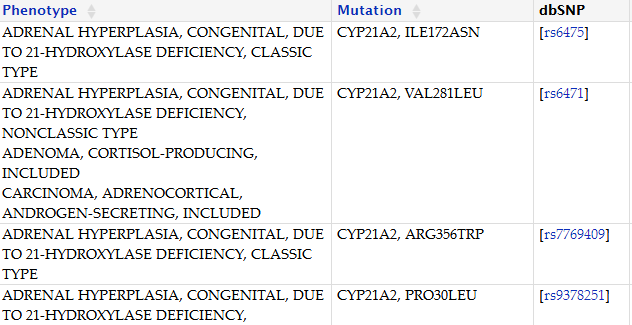
1、21-羟化酶缺乏症
21-羟化酶缺乏症是常染色体隐性遗传疾病,表现为先天性肾上腺皮质增生,属于肾脏疾病系统。
致病原因:该病是由于CYP21基因突变,导致21羟化酶缺乏。进而造成糖皮质和(或)盐皮质类固醇减少,ACTH和雄激素分泌增多,患者出现男性化和失盐症状,严重时威胁生命。
临床表现:拒食、呕吐、脱水、体质量不增或下降及电解质紊乱、嗜睡为表现的肾上腺危象及外生殖器两性畸形、患儿性早熟、男性化体征等。
致病基因:CYP21A2
2、白化病(Albinism)
白化病一般表现为常染色体隐性遗传, 属于皮肤疾病系统。
致病原因:该疾病是一组由黑色素合成相关的基因突变导致眼或眼、皮肤、毛发黑色素缺乏引起的遗传性疾病。
致病基因:TYR,OCA2,SLC45A2,GPR143,TYBP1,SLC24A5
临床表现:主要表现为眼、皮肤、毛发黑色素缺乏和弱视等。患者全身皮肤呈白色或粉红色,毛发呈白色或淡黄色,具有视力低、畏光、眼球震颤等表现。
基因突变位点:


3、Alport综合征
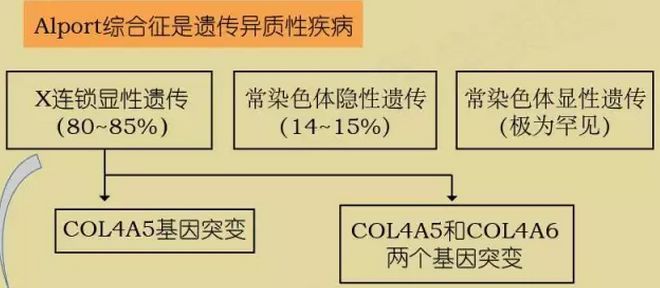
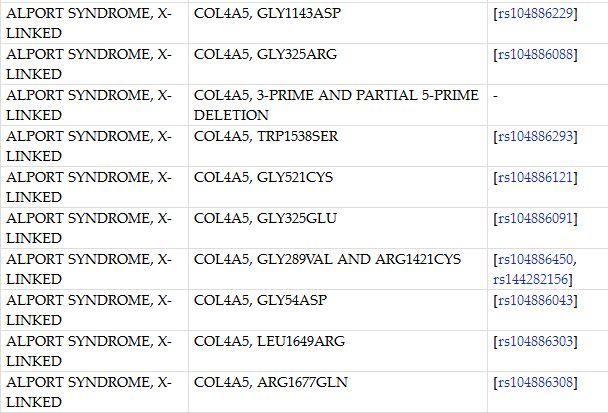
Alport综合征(Alport syndrome, AS)是以进行性血尿, 肾功能不全为主, 伴有耳聋和/或眼病变的一种遗传性疾病。该疾病属于肾脏疾病,本病主要遗传方式是x-连锁显性遗传。男性比女性发病早且严重, 可因肾功能衰竭而死亡。
致病基因:COL4A5,COL4A6,COL4A3/COL4A4
临床表现:
肾脏症状:腰痛、间歇性或持续性血尿。
肾外症状:感音神经性耳聋:进行性,两侧不完全对称;晶状体异常,进行性近视甚至白内障;视网膜异常染色;高血压;水肿。
基因突变位点:
4、肌萎缩侧索硬化(ALS)
肌萎缩侧索硬化症(ALS)是既累及上运动神经元(UMN),又影响到下运动神经元(LMN)及其支配的躯干、四肢和头面部肌肉的一种慢性进行性变性疾病。
ALS俗称「渐冻人症」,因「冰桶挑战」项目广为人知。

临床表现:肌无力、肌萎缩、肌束震颤,延髓麻痹和锥体束征等。
致病基因:SETX
基因变异位点:

相关治疗报道:
NEJM:苯丁酸钠-牛磺二醇治疗肌萎缩性侧索硬化症的疗效分析
Orphazyme的热休克蛋白疗法Arimoclomol治疗肌萎缩性侧索硬化症,获得美国快速通道指定
Neuropore宣布其NPT520-34治疗肌萎缩性脊髓侧索硬化症,喜获FDA的孤儿药物指定
Cytokinetics的快速骨骼肌肌钙蛋白激活剂Reldesemtiv获得欧洲孤儿药称号,用于治疗肌萎缩性侧索硬化症
5、苯丙酮尿症
苯丙酮尿症(Phenylketonuria,PKU)是一种先天性代谢性疾病,由于苯丙氨酸代谢途径中的酶缺陷,使得苯丙氨酸不能转变成为酪氨酸,导致苯丙氨酸及其酮酸蓄积并从尿中大量排出。临床主要表现为智能低下,惊厥发作和色素减少。本病属常染色体隐性遗传。其发病率随种族而异,美国约为1/14000,日本1/60000,我国1/16500。若不及时治疗,会对脑或神经系统造成不同程度的损害,给患者带来终身痛苦甚至生命威胁。多数患者在婴儿期便会出现临床症状,并随年龄增大而加重。由于患者无法转化普通含蛋白质食物中的苯丙氨酸,所以平常的肉、鱼、蛋、奶、豆等含蛋白质的食物摄取要严格控制。目前对PKU的治疗,大多以食疗为主。
相关报道:欧盟批准Palynziq用于治疗苯丙酮尿症成年患者
6、瓷娃娃--成骨不全症
成骨不全症(Osteogenesis imperfecta,OI)又称脆骨症,发病概率在一万分之一到一万五千分之一左右,中国约有10万患者,是一种显性遗传的疼痛疾病。该疾病目前分为八型,普遍表现为骨脆弱和骨畸形,严重者轻轻一个拥抱也会导致其骨折,该群体如不及时治疗将终生残疾。主要特征为:频繁骨折、身材矮小、听力渐进性失聪、眼睛巩膜呈蓝色等,由于患者极易骨折,民间俗称"瓷娃娃",尚无治愈方法,但通过药物治疗和矫形手术可以显著提高患者生存质量。
相关报道:2019 ASBMR:硬化蛋白单抗Setrusumab在成骨不全症方面有治疗潜力
7、玻璃娃娃--血友病
血友病(Hemophilia),是一组由于血液中某些凝血因子的缺乏而导致患者产生严重凝血障碍的遗传性出血性疾病,男女均可发病,但绝大部分患者为男性。包括血友病A(甲)、血友病B(乙)和因子XI缺乏症(曾称血友病丙)。前两者为性连锁隐性遗传,后者为常染色体不完全隐性遗传。血友病在先天性出血性疾病中最为常见,出血是该病的主要临床表现。由于血友病患者受到磕碰或者创伤后容易引起淤血或者血流不止,因此他们被称作碰不得摔不得的"玻璃娃娃"。
8、淋巴管肌瘤病
淋巴管肌瘤病(Lymphangioleiomyomatosis, LAM)是一种罕见的疾病,几乎所有的病例均发生于女性,以育龄期女性为主,平均年龄30-40岁。气胸和乳糜胸常为LAM的首发症状,并可反复发生。随着疾病的发展,会出现呼吸困难并进行性加重,最后可出现较为严重的呼吸衰竭。目前在我国报告病例数不超过200例。LAM患者大部分处于承担家庭和社会责任的年龄,却被疾患所困,失去了正常的工作和生活。
9、特发性肺动脉高压
特发性肺动脉高压(idiopathic pulmonary arterial hypertension,IPAH)它是指经详细体格检查及完善实验室检查均未发现引起肺动脉高压的其他系统疾病及相关线索,并且经右心导管检查发现肺毛细血管嵌压<20mmHg。根据美国研究资料,年发病率约为1~2 / 1,000,000,女性发病率高于男性,比例约为2~3∶1。特发性肺动脉高压是一极度恶性疾病,其自然病程较短,患者往往病情发展快,如无正确治疗,很快会死于难以纠正的右心衰竭,平均生存时间为2~3年,其治疗困难,预后差,严重程度甚至远远超过了癌症,被医生称为“心脏病中的癌症”。肺动脉高压虽然目前尚无法治愈,但患者通过正规的治疗和靶向药物的治疗能够有效地改善生存质量。
10、克氏综合症
克氏综合症(Klinefelter's syndrome)是一种性染色体异常引起的遗传疾病,其个体有至少2条XX性染色体,至少1条Y性染色体,典型情况下为XXY,一般为男性。该症于1942年首先由 Klinefelter 发现并描述。额外的X染色体,可能来自卵子或精子。发病率为0.1%。克氏综合症本身并不能致人死亡,导致病患身心困扰的都是并发症和并生症所致。其常见并生症和并发症包括:乳腺增生,骨质疏松,二型糖尿病,系统性红斑狼疮,自闭症,乳腺癌等等。
11、结节性硬化症
结节性硬化症(Tuberous Sclerosis Complex,TSC)是一种多系统受累的常染色体显性遗传病,儿童和成人均可受累。主要表现为癫痫、皮肤病变、多器官良性肿瘤,部分病人智力发育受影响,有1/3的女性患者肺部出现淋巴管肌瘤病。TSC的基因突变发生在TSC1或TSC2,导致了细胞增长失控。TSC无法在怀孕期间检测出来,大多数患者在发病前都毫无前兆,也不可能预测病患将如何发展,和将会有哪些症状。结节性硬化症无论给患者本人还是其家庭都带来了巨大的心理压力,非常需要更多的社会理解和关注、定期的医疗检查、专业的心理辅导,去帮助他们踏上这趟人生的不确定之旅。
12、Angelman氏症候群
天使人综合症(Angelman syndrome),或称天使综合症,又称安格曼综合症,是一种基因缺陷而造成的疾病。罹患此症的患者,脸上常有笑容,缺乏语言能力、过动,且智能低下。
临床表现:患者脸上始终带着笑容,但动作机械,智力低下,同时还有癫痫等症状。病征包身边经常发笑,双手举高,挥舞,脚下不稳,痉挛,缺乏语言能力及智障,因此被称为"快乐木偶综合症"。
相关报道:天使人综合征的治疗迎来新突破!FDA授予OV101孤儿药称号
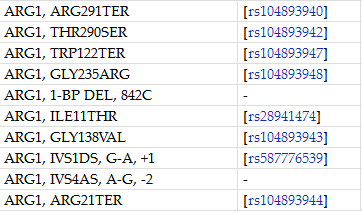
13、精氨酸酶缺乏症
精氨酸酶缺乏症:属常染色体隐性遗传病,属于内分泌与代谢疾病系统。
致病原因:由于ARG1基因缺陷导致肝脏精氨酸酶缺乏,精氨酸降解障碍,鸟氨酸与尿素生成减少,血液氨含量增高,是先天性尿素循环障碍中较少见的类型。该病多发于婴幼儿(1-3岁)。
临床表现:患者临床常表现为生长发育迟缓、痉挛性瘫痪、癫痫及小脑萎缩,早期常常被误诊为脑性瘫痪,生存质量极差,预后不良。
致病基因:ARG1
ARG1基因突变位点:
14、热纳综合征(窒息性胸腔失养症)
窒息性胸腔失养症是一种罕见的遗传性骨骼生长障碍,其主要影响胸部骨骼结构的发育,导致畸形。
临床表现:常伴有呼吸窘迫和反复呼吸道感染。
非典型溶血性尿毒症
非典型溶血尿毒综合征属于肾脏疾病系统。是一种补体失调性疾病,补体调控蛋白H因子、以及膜辅助蛋白和血清补体固有成分(B因子、补体C3)的基因突变都可参与其发病,病情易反复,预后很差,25%的患者在急性期死亡,50%以上发展为终末期肾病。
临床表现:最主要特点有,微血管溶血性贫血、血小板减少及肾功能衰竭。
致病基因:HF1,CFHR3,CFHR1
基因突变位点:
CFHR3基因:84-KB DEL
CFHR1基因:84-KB DEL
15、自身免疫性脑炎
AE泛指一大类由于免疫系统针对中枢神经系统抗原产生反应而导致的疾病,是非感染性因素所致可逆性脑炎。


临床表现:
(1)精神症状与神经系统症状常同时出现,或神经系统阳性体征出现稍晚;
(2)兴奋、话多、行为异常等精神运动性兴奋;
(3)情感淡漠、反应迟钝、少言少语等精神运动性抑制、亚木僵状态、幻觉及被害妄想、忆力下降、头痛、发热 ,癫痫样发作、言语及运动障碍、自主神经功能紊乱(多汗)等。
16、自身免疫性垂体炎
自身免疫性垂体炎是由于自身免疫引起的脑垂体慢性炎症,也被称为淋巴细胞性垂体炎。
临床表现:
(1)妊娠时垂体发生生理性肿大;
(2)垂体体积增大、头痛、视力障碍、视野缺损等;
(3)垂体前叶功能异常;
(4)血清垂体自身抗体的检出及其它内分泌腺的受累。
注意:头疼是该疾病最常见的首发症状。
17、自身免疫性胰岛素受体病
自身免疫性胰岛素受体病是一种由胰岛素受体的自身抗体引起的罕见疾病。
临床表现:系统性红斑狼疮是该疾病最常见的并发症。
18、戈谢病(Gaucher disease, GD)
戈谢病是由GBA酸性葡萄糖苷酶(acid mai -glucosidase, GBA)基因突变引起的一种最常见的遗传性溶酶体储存疾病。本研究旨在确定GD突变及其基因型-表型的相关性。
Prevail Therapeutics是一家生物技术公司,致力于为神经退行性疾病患者开发基于腺病毒(AAV)的基因疗法,该公司近日宣布,美国FDA已通知Prevail,PR001用于治疗神经性戈谢病(nGD)的IND已生效,并且Prevail可能会着手进行临床试验。
戈谢病#相关话题
相关治疗药物报道:基因疗法PR001治疗神经性戈谢病:IND已生效
19、法布雷病
法布雷病(Fabry disease)又称“Anderson-Fabry 病”(Anderson-Fabry disease, AFD),1898 年分别由两位皮肤科医师William Anderson(德国)和Johannes Fabry(英国)最早报道,由此得名。该病是一种罕见的X染色体伴性遗传的溶酶体贮积病(lysosomal storage diseases,LSDs)。由于编码α-半乳糖苷酶A(α-Gal A)的基因发生突变,导致该酶活性部分或全部丧失,造成其代谢底物三己糖酰基鞘脂醇(Gb3)和相关的鞘糖脂在人体各器官大量沉积,最终引起一系列脏器病变。
由于编码α-半乳糖苷酶A(α-Gal A)的基因发生突变,导致该酶活性部分或全部丧失,造成其代谢底物三己糖酰基鞘脂醇(Gb3)和相关的鞘糖脂在人体各器官如心脏、肾脏、胰腺、皮肤、肺和神经系统等大量贮积,最终引起一系列脏器病变。
话题:#法布雷病#
相关治疗药物报道:又一种罕见病药物国内上市,全国仅约300名患者
20、杜氏肌肉营养不良症( Duchenne Muscular Dystrophy,DMD)
杜氏肌肉营养不良症乃遗传性肌肉萎缩病。它的基因( Dystrophin gene)存在于X性染色体中( Xp21 ),因此它是透过性连锁式隐性遗传型态传播的。男性只有一个X性染色体,因此病患者大多为男性;若女性的一对X性染色体中其一个携有异变的 Dystrophin 基因,她便成为一个 DMD 的携带者,她的儿子有二分一的机会成为病患者,她的女儿则有二分一机会成为 DMD基因携带者。
相关治疗药物报道:
Edasalonexent治疗杜兴氏肌营养不良症的III期PolarisDMD试验即将开始
杜氏肌营养不良新药Viltolarsen下月申请,有望获批,本药在2020年已在美国上市
21、肝豆状核变性
肝豆状核变性(hepatolenticular degeneration,HLD)由Wilson在1912年首先描述,故又称为Wilson病(Wilson Disease,WD)。是一种常染色体隐性遗传的铜代谢障碍性疾病,以铜代谢障碍引起的肝硬化、基底节损害为主的脑变性疾病为特点,对肝豆状核变性发病机制的认识已深入到分子水平。WD的世界范围发病率为1/30 000~1/100 000,致病基因携带者约为1/90。本病在中国较多见。WD好发于青少年,男性比女性稍多,如不恰当治疗将会致残甚至死亡。WD也是至今少数几种可治的神经遗传病之一,关键是早发现、早诊断、早治疗。
22、亨廷顿舞蹈症(Huntington’s disease, HD)
亨廷顿舞蹈症是一种罕见的常染色体显性遗传病。患者一般在中年发病,出现运动、认知和精神方面的症状。亨廷顿舞蹈症临床症状复杂多变,患者病情呈进行性恶化,通常在发病 15~20 年后死亡。起病隐匿,进展缓慢,以舞蹈样动作伴进行性认知、精神功能障碍终至痴呆为该病的主要特征。病因是亨廷顿基因上多核苷酸重复序列的错误表达,从而影响不同的分子通路,最终导致神经功能失调和退化。
相关治疗药物报道:氘代丁苯那嗪在中国申报上市治疗亨廷顿舞蹈症(HD)
23、特发性肺纤维化(IPF)
特发性肺纤维化(IPF)是一种慢性、进行性、纤维化性间质性肺疾病,病变局限在肺脏,好发于中老年人群,其肺组织学和/或胸部高分辨率CT(HRCT)特征性表现为普通型间质性肺炎(UIP),病因不清。按病程有急性、亚急性和慢性之分,本病多为散发,据统计,每年整体人群中的患病率约(2~29)/10万,且呈逐渐增长趋势,估计以每年11%的比例增长。在美国特发性肺纤维化患者大约有100000人,欧盟地区大约有110000人,而且每年欧盟地区新增IPF患者35000人。日本每年整体人群中的IPF患病率约(2.23~10)/10万,实际值远高于这个数目。我国作为一个老龄化严重的国家,目前IPF患病人数也是逐年增加,保守估计至少在50万左右。作为一种慢性间质性肺病,IPF起病隐匿、病情逐渐加重,也可表现为急性加重。IPF诊断后的平均生存期仅2.8年,死亡率高于大多数肿瘤,IPF被称为一种“类肿瘤疾病”。
报道:
特发性肺纤维化的治疗突破!AT2R激动剂VP01或具强大作用
Eur Respir J:吡非尼酮与吸入N-乙酰半胱氨酸联合治疗特发性肺纤维化疗效
24、Leber遗传性视神经病变
Leber遗传性视神经病变(Leber′s hereditary optic neuropathy,LHON)属母系遗传(maternal inheritance)或称线粒体遗传,由线粒体DNA11778、14484或3460等位点突变引起。它是遗传性视神经病变的常见类型,由德国学者Leber于1871年首先报道。目前国外正在开发相关的基因治疗药物,见:基因疗法NR082治疗Leber遗传性视神经病变:FDA已授予孤儿药称号(ODD),Leber遗传性视神经病变的治疗或许不再是难题:GS010的REVERSE III期试验数据即将公布
25、枫糖尿症
枫糖尿症(MSUD)又称槭糖尿病、支链酮酸尿症,是一种常染色体隐性遗传病,由于分支酮酸脱羧酶的先天性缺陷,致使分支氨基酸分解代谢受阻,因患儿尿液中排出大量α-酮-β-甲基戊酸,带有枫糖浆的香甜气味而得名。
26、黏多糖贮积症
黏多糖贮积症是由于人体细胞的溶酶体内降解黏多糖的水解酶发生突变导致其活性丧失,黏多糖不能被降解代谢,最终贮积在体内而发生的疾病。该病是溶酶体贮积病中非常重要的一类,可分为Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅵ,Ⅶ,Ⅸ型等7种型,其中Ⅲ又分为ⅢA,ⅢB,ⅢC,ⅢD四个亚型,Ⅳ型分为ⅣA和ⅣB亚型,虽然各型致病基因和临床表现有差异,但由于贮积的底物都是黏多糖而被统称为黏多糖贮积症。
相关治疗报道:黏多糖贮积症(Mucopolysaccharidsis,MPS)基因治疗计划取得积极中期数据
自体造血干细胞基因疗法OTL-203治疗I型黏多糖贮积症:获得FDA的孤儿药和罕见儿科疾病称号
赛诺菲携3款罕见病新药亮相进博会: 为法布雷病、血友病及黏多糖贮积症患者带来新希望
27、尼曼-匹克病(NPD)
尼曼-匹克病(NPD)又称鞘磷脂沉积病,属先天性糖脂代谢性疾病。其特点是全单核巨噬细胞和神经系统有大量的含有神经鞘磷脂的泡沫细胞。为常染色体隐性遗传,目前至少有五种类型。
28、肝卟啉病
29、肾上腺脑白质营养不良
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是一种致命性、神经退行性、X连锁隐性遗传病,发病率 0.5/10 万~1/10 万,多见于年轻男孩儿,影响全球大约21000例男性新生儿。
ALD发病原因是ABCD1 基因突变导致其编码的肾上腺脑白质营养不良蛋白(ALDP)结构改变或功能缺失,造成患者体内的极长链饱和脂肪酸(VLCFA) 不能进入过氧化物酶体进行β-氧化, 从而在血浆和组织细胞中蓄积,导致线粒体功能障碍,氧化应激加剧,生物膜功能破坏,最终引起神经系统脱髓鞘和肾上腺皮质功能减退等病理变化。
相关治疗报道:改善致死罕见病CALD患者长期生存结局
30、真性红细胞增多症
真性红细胞增多症(polycythemia vera, PV)是一种骨髓增殖性肿瘤,骨髓增殖性肿瘤是一组源于造血干细胞、以形态学成熟度不一的骨髓细胞克隆增殖为特征的恶性肿瘤。PV与其他MPN的区别在于红细胞量增多,且与血栓栓塞事件、白血病转化和/或骨髓纤维化的风险增加有关。
Protagonist制药公司今日宣布,美国食品药品监督管理局(FDA)已授予PTG-300治疗PV的孤儿药物认定。PTG-300是一种合成多肽,目前正在临床开发中。见:关注罕见病“真性红细胞增多症”:PTG-300获美国FDA孤儿药物认定
31、地中海贫血症
地中海贫血(Thalassemias)是遗传性血液疾病,会造成血红蛋白合成障碍,其症状可依不同分型而有所不同,程度可能从无症状到严重。通常地中海贫血伴随典型的贫血症状,即红血球细胞水平低下。贫血可导致疲累感与肤色苍白,也可同时造成骨骼疾病、脾脏肿大、黄疸、深色尿以及儿童成长迟缓等症状。见:关注罕见病“地中海贫血症”:FDA授予Mitapivat孤儿药称号
32、红斑性肢痛症
红斑性肢痛症(erythromelalgia)是一种原因不明的肢端远端皮肤阵发性皮温升高,皮肤潮红、肿胀,并产生剧烈灼热痛为特征的一种植物神经系统疾病。环境温度升高可诱发或加剧疼痛;温度降低可使疼痛缓解。任何年龄均可起病,但以青壮年多见。目前也有新药在研发,见:关注罕见病“红斑性肢痛症”:FDA授予ATX01孤儿药称号
33、转甲状腺素蛋白淀粉样变性心肌病(ATTR-CM)
ATTR-CM是一种罕见的致死性疾病,由一种转运蛋白,转甲状腺素蛋白不稳定引起的。转甲状腺素蛋白由4个相同的部分(四聚体)组成。当不稳定的转甲状腺素蛋白四聚物解离,就会导致错误折叠的蛋白形成淀粉样纤维沉积于心脏,从而使心肌变得僵硬,最终导致心力衰竭。相关治疗药物报道:罕见病新药!辉瑞「氯苯唑酸软胶囊」在中国获批
34、α1-抗胰蛋白酶缺乏症
α1-抗胰蛋白酶缺乏症是血中抗蛋白酶成分-α1-抗胰蛋白酶(简称α1-AT)缺乏引起的一种先天性代谢病,通过常染色体遗传。临床常导致新生儿肝炎,婴幼儿和成人的肝硬化、肝癌和肺气肿等。相关报道:α-1抗胰蛋白酶(A1AT)缺乏症治疗或取得重大突破:DCR-A1AT获得孤儿药认定
35、脊髓性肌萎缩症(SMA)
脊髓性肌萎缩症(SMA),是一类由脊髓前角运动神经元变性导致肌无力、肌萎缩的疾病。属常染色体隐性遗传病,临床并不少见。本病临床表现差异较大,根据患者起病年龄和临床病程,将SMA由重到轻分为4型。共同特点是脊髓前角细胞变性,临床表现为进行性、对称性,肢体近端为主的广泛性弛缓性麻痹与肌萎缩,智力发育及感觉均正常。
治疗报道:
罗氏的risdiplam在1型脊髓性肌萎缩症婴儿中展现出治疗潜力
诺华的基因疗法Zolgensma,在日本获批治疗脊髓性肌萎缩症(SMA)儿童患者
36、重症肌无力
重症肌无力(MG)是一种由神经-肌肉接头处传递功能障碍所引起的自身免疫性疾病,临床主要表现为部分或全身骨骼肌无力和易疲劳,活动后症状加重,经休息后症状减轻。患病率为77~150/100万,年发病率为4~11/100万。女性患病率大于男性,约3:2,各年龄段均有发病,儿童1~5岁居多。
相关治疗报道:
抗体片段efgartigimod治疗重症肌无力:III期试验迎硕果!
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#收藏#
129
#罕见#
91
赶紧mark,收藏啊,everybody
103
关注罕见病
111
#罕见病#这是目前看到最经典,最全面的信息,收藏!!
171