
COVID-19“快乐性”低氧血症的病理生理学
2023-03-31 刘岗 重症医学 发表于上海

本综述描述了COVID-19的病理生理学异常,这可能解释了为何患者严重低氧血症,却只有相对轻度的呼吸不适。
摘要
新型冠状病毒肺炎(COVID-19)大流行是一场全球性危机,对全球医疗系统构成重大挑战。许多患者在静息状态下有严重低氧血症,并可能快速恶化,但无相应的呼吸窘迫体征(即快乐性低氧血症),表现出明显的脱节。COVID-19患者的这种特殊临床表现与医生通常治疗呼吸衰竭重症患者的经验截然不同,因此确保及时将该类患者转诊至重症监护室可能具有挑战性。彻底了解呼吸驱动和低氧血症的病理生理学决定因素可能有助于更全面地理解患者的临床表现和管理。尽管动脉血样中氧分压较低,但由于低氧血症驱动的过度通气诱导氧合血红蛋白解离曲线左移以及病毒可能与血红蛋白的直接相互作用,导致动脉血氧饱和度不变。通气-血流不匹配(从分流到肺泡无效腔通气)是中心标志,这些病理生理学提供各种治疗靶点。
关键信息
本综述描述了COVID-19的病理生理学异常,这可能解释了为何患者严重低氧血症,却只有相对轻度的呼吸不适。
背景
2019年12月初,湖北省省会武汉发现首例不明原因肺炎病例。由于与SARS-CoV和中东呼吸综合征(MERS)病毒相似,新型冠状病毒肺炎(coronavirus disease 2019,COVID-19)的病原被鉴定为包膜RNA乙型冠状病毒属家族的新成员,并命名为重度急性呼吸综合征冠状病毒2型(SARS-CoV-2)。尽管对COVID-19的流行病学和临床特征了解较多,但对其对肺病理生理学的影响知之甚少。COVID-19的临床严重程度变异性大,数据将病例分类为轻度(81%)、重度(14%)或危重(5%)。许多患者有明显的动脉低氧血症,但无成比例的呼吸窘迫体征,甚至未述说呼吸困难感。该现象被称为沉默或“快乐”低氧血症。Tobin等人最近报告了3例快乐低氧血症,PaO2为36-45 mmHg(PaCO2为34-41 mmHg),肺泡通气未增加。在COVID-19患者中,低氧血症的严重程度与住院死亡率独立相关,可成为患者入住重症监护室(ICU)风险的重要预测因素。由于正确识别低氧血症对预后和及时的治疗决策有影响,我们在此概述了COVID-19的病理生理学异常,这可能解释了为何低氧血症和患者呼吸困难感觉间有脱节。
呼吸困难感
呼吸受脑干延髓和脑桥呼吸中枢集中控制(见图1),呼吸中枢控制“呼吸驱动”,使呼吸与身体的代谢需求相匹配。呼吸驱动主要来自外周和中枢化学感受器间的化学反馈,但呼吸中枢也受到高级大脑皮层、下丘脑综合伤害性感受、肌肉和肺机械牵张受体的反馈以及代谢率的影响。呼吸中枢的输出信号可分为节律(如呼吸频率)和模式(如呼吸用力深度)信号,这些输出信号可独立控制。呼吸困难一般定义为感觉“不适、困难或费力”,一般情况下当通气需求与患者的反应能力不成比例时出现呼吸困难。应与呼吸急促(呼吸频率增加)或呼吸过度(通气增加)相区别。呼吸困难分级与这种感觉是发生在静息还是运动时相关。这种半定量评分方法的最佳例证是经常使用的改良医学研究委员会(MRC)呼吸困难量表,相对于同龄受试者,该量表将呼吸困难从0级(仅剧烈运动时呼吸困难)分类到4级(因呼吸困难而无法出门或穿衣时呼吸急促)。
通过大脑皮层和下丘脑,各种感觉、疼痛和情绪刺激影响呼吸感。肌肉运动的感觉异常是导致呼吸困难的另一因素。健康呼吸中,对呼吸肌激活是无意识的。然而,当呼吸肌由于肺力学改变(例如,胸廓顺应性降低)而疲劳或减弱时,呼吸可视为一种需要付出巨大努力的动作。呼吸道和胸壁的机械感受器输入也可引起呼吸困难。刺激迷走神经受体(如支气管收缩、呼吸时施加外部阻力)似乎会加重呼吸困难。尽管代谢率在运动期间呼吸困难中的作用已得到确认,但在调节重症患者呼吸困难感觉中的作用仍不清楚。呼吸驱动最广为人知的决定因素是中枢和外周化学感受器。可导致外周和中枢化学感受器水平pH值变化的血液中溶解二氧化碳的血气分压(PaCO2)变化,似乎是最重要的影响因素。稳态时,动脉PaCO2由以下公式确定:


高碳酸血症(由VD增加、通气不足或VCO2增加引起)的正常反应是呼吸驱动和每分钟通气量增加。与高碳酸血症本身能导致呼吸困难不同,低氧血症本身在心肺疾病患者经历的呼吸急促感觉中的作用有限。在健康受试者中,轻度低氧血症(PaO2 60-65mmHg)(例如停留在高海拔或实验性低氧室所致)时,呼吸驱动增加极小。许多呼吸困难的患者没有低氧血症,而那些有低氧血症的呼吸困难患者,经过氧疗纠正低氧血症后呼吸困难的症状仅获得轻微改善。当动脉PaO2<40 mmHg时,常出现呼吸困难。值得注意的是,对低氧血症的正常反应是每分钟通气量的增加,主要是通过增加潮气量和呼吸频率。因此,不是呼吸困难,而是呼吸率(呼吸急促)和潮气量增加(呼吸过强)是即将发生低氧性呼吸衰竭的最重要临床体征。此外,PaCO2是调节脑血流的基本因子之一。过度通气降低PaCO2,随后导致动脉血管收缩,从而降低脑血流量和颅内压。相反,PaCO2升高导致颅内压升高,最终恶化意识水平、脑干反射改变以及姿势和运动反应改变。在床旁,深入了解呼吸驱动和低氧血症的病理生理学决定因素可促进对COVID-19临床表现的更全面理解和及时处理。
COVID-19 快乐低氧血症
COVID-19患者的重度低氧血症,而报告的呼吸不适相对轻度,这与医生通常治疗呼吸衰竭重症患者获得的经验形成对比。在1099例住院的COVID-19患者中,尽管PaO2/FiO2比值较低、CT扫描异常(86%)和通常需要氧疗(41%),Guan报告仅18.7%的患者出现呼吸困难。快乐或无症状的低氧血症并不仅见于COVID-19,也可发生于肺不张、肺内分流(即动静脉畸形)或右向左心内分流的患者。气体交换是否充分主要由肺通气和毛细血管血流之间的平衡决定,称为通气/血流(V/Q)匹配。在COVID-19的初始阶段,几种机制会造成动脉低氧血症(见图2),但此时呼吸功不增加,低氧血症可能会快速恶化。
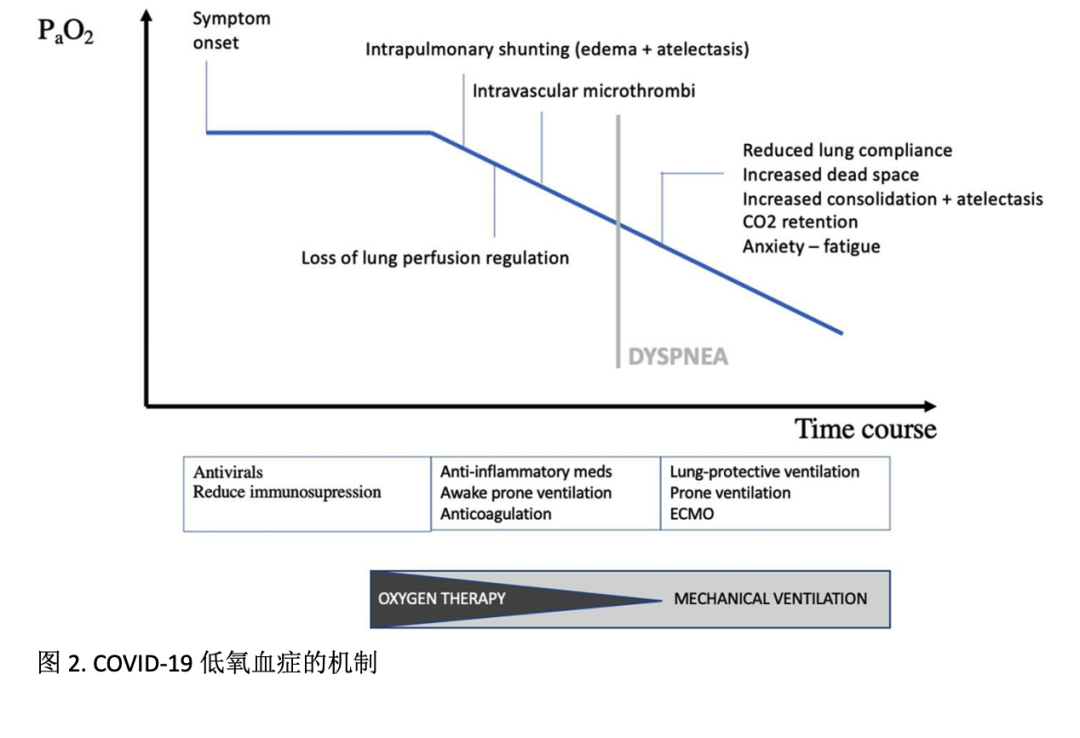
氧合血红蛋白解离曲线的变化
脉搏血氧饱和度(SpO2)通常用于检测低氧血症。但在COVID-19中,SpO2应谨慎解读。由于低氧血症驱动的呼吸急促和呼吸过度,诱导呼吸性碱中毒(PaCO2下降),S形氧合血红蛋白解离曲线似乎左移。在低碳酸血症期间,对于给定程度的PaO2,血红蛋白与氧的亲和力增加,从而氧饱和度增加,这就解释了为什么面对极低的PaO2,可以很好地维持SpO2在正常水平。这一发现也见于高原低氧血症,高原时的低碳酸血症可显著改变氧-血红蛋白解离曲线,改善血氧饱和度。肺泡气体方程还可预测过度通气以及由此引起的肺泡二氧化碳分压下降可导致肺泡氧分压增加,并最终升高SpO2。
COVID-19 曲线左移也可能有生物学解释。Liu等人提出了关于病毒与血红蛋白中血红素基团直接相互作用的假说。根据这一理论,COVID-19患者血清血红素水平随着有害铁离子(Fe3+) 的增加而升高,导致炎症和细胞死亡(铁死亡)。为减少组织损伤,产生大量血清铁蛋白结合这些游离铁。总之,应根据是否有过度通气(呼吸急促、低PaCO2)、若可以应通过动脉PaO2,来解释SpO2。计算肺泡-动脉血氧梯度(P(A-a)O2梯度=(150 mmHg-PaCO2/0.8-海平面PaO2),并考虑年龄(年龄/4 + 4 + 50(FiO2–0.21),单位mmHg)和氧疗等影响因素,可能有意义。计算肺泡-动脉血氧梯度可在智能手机的应用程序上快速执行。V/Q不匹配或肺内分流可增加 P(A-a)O2 梯度。V/Q不匹配导致的低氧血症可通过氧疗轻松纠正,而氧疗对肺分流效果较差。
COVID-19 低氧血症的原因
肺内分流
SARS-CoV-2感染早期的动脉低氧血症主要是由V/Q不匹配引起的,因此肺动脉血流持续流到非通气的肺泡,表现为P(A-a)O2梯度显著增加。感染可导致局部间质轻度水肿,尤其是在应力和应变集中的、不同弹性性质的肺结构间的界面处。由于肺水肿增加(导致胸部成像中毛玻璃样阴影和实变)、表面活性物质丢失和叠加压力,随后发生肺泡塌陷,相当一部分心输出量灌注非通气肺组织,导致肺内分流。如前所述,发病时潮气量增加,导致吸气性胸腔内负压升高。正如Barach在1938年首次描述的,胸腔内负压与炎症导致的肺通透性增加相结合,最终导致进行性水肿、肺泡溢出和患者自戕性肺损伤(P-SILI)。随着时间的推移,水肿增加将进一步增强肺重量、肺泡萎陷和重力依赖性区的肺不张,导致分流量逐渐增加和氧合进一步下降,通过增加FiO2无法完全纠正低氧血症。
肺血流调节丧失
正如Lang等人最近使用双能量CT所显示的那样,SARS-CoV-2感染期间低氧性肺血管收缩机制(小肺内动脉对肺泡缺氧的反应而收缩)相对失效似乎是高肺血流量持续至非通气肺泡的原因。低氧性肺血管收缩机制相对失效是否仅由内源性血管扩张剂(前列腺素、缓激肽和与炎症过程相关的细胞因子释放)或由其他尚未确定的机制触发,仍有待研究。作为P-SILI作用机制的一部分,可能由肺结构间界面的剪切应力诱导的血管麻痹似乎也损害了肺血流调节。此外,肾素-血管紧张素系统(RAS)失调参与了COVID-19的病理生理学。血管紧张素转换酶2受体(ACE2)是SARS CoV-2进入细胞的主要功能受体,意味着ACE2对SARS CoV-2的内化作用。ACE2将血管紧张素Ⅱ(AngⅡ)转化为血管紧张素1-7(Ang 1-7),对降解缓激肽也有重要意义。因此,降低的ACE2水平导致Ang Ⅱ升高,通过激动Ang Ⅱ受体介导肺血管收缩,而Ang 1-7拮抗AngⅡ的作用。最近,Liu等人发现,血清AngⅡ水平与COVID-19的病毒载量和肺损伤线性相关。
血管内微血栓
内皮损伤正在成为COVID-19发病机制的中心标志,细胞致病病毒可直接感染表达ACE2的肺毛细血管内皮细胞。血管内微血栓是急性炎症和内皮损伤时促凝和纤溶活性失衡的净效应。促凝活性可能是由补体系统介导的凝血激活(类似于某些血栓性微血管病(TMA)),也可能是由于纤溶酶原激活物抑制剂(PAI-1和-2)的活性增加抑制了纤溶酶原激活和纤溶(在IL-6 的影响下,PAI-1和-2作为急性期蛋白被诱导)。通过内皮释放组织因子和激活凝血因子VII和XI,重度COVID-19 患者也可能有弥散性血管内凝血(DIC)。许多COVID-19 患者D-二聚体升高,提示血栓形成。入院时的D-二聚体水平用于预测COVID-19的住院死亡率,预后不佳的COVID-19患者中,DIC的频率更高(71%),而存活者中仅为0.6%。重症COVID-19的肺尸检显示纤维蛋白沉积、弥漫性肺泡损伤、血管壁增厚,常出现富含补体的微血栓阻塞肺毛细血管和较大血栓引起肺动脉血栓和栓塞。高凝状态进一步恶化V/Q不匹配和肺组织损伤。此外,作为COVID-19急性期蛋白,活化C反应蛋白和随后的补体激活以及肝脏合成的纤维蛋白原也可调节凝血功能。
弥散功能受损
肺弥散量(DLCO)可能受损,尽管纯弥散障碍很少导致静息时P(A-a)O2梯度增加。SARS-CoV2在肺泡II型细胞内繁殖,在此过程中会产生并释放大量病毒颗粒,随后发生免疫应答介导的感染细胞破坏(病毒相关性焦亡)。肺泡上皮细胞丢失和促凝血状态导致裸露的基底膜被透明膜(由纤维蛋白、死亡细胞和补体活化产物组成)覆盖。随着运动量增加和COVID-19缺氧性肺血管收缩的缺失,高动力肺循环可能使红细胞没有足够的时间平衡其摄氧量。因此,COVID-19可能出现弥散受限,导致P(A-a)O2梯度升高和运动诱发的动脉低氧血症(EIAH)。最近,Xiaoneng Mo等人证实COVID-19患者出院时DLCO降低。弥散功能受损的患病率与疾病严重程度相关,轻症疾病、肺炎和重症肺炎分别为30.4%、42.4%和84.2%。需要长期研究来阐明这些功能障碍是否如在中东呼吸综合征中所见的那样是持续的,37%的中东呼吸综合征幸存者有DLCO受损。
肺力学基本完整
前面段落中的概述基本澄清了COVID-19中严重的低氧血症与相对完好的肺力学间的脱节。某些COVID-19患者的气体交换异常发生时间早于机械负荷的增加。在感染的前几天,气道阻力没有增加,推测解剖或生理无效腔通气没有增加。呼吸困难也仍然相当低,因为在许多无既存肺病的患者中,肺顺应性正常。正如Gattinoni等最近在16例重症患者队列所显示的,相对正常的呼吸系统顺应性(50.2±14.3 mL/cmH2O)与分流率(0.50±0.11)的急剧增加同时出现。无论是导致急性肺损伤还是ARDS的大多数疾病,如此大的脱节非常罕见。相对较高的顺应性表明对肺通气量影响较小,一定程度上解释了疾病早期无呼吸困难的原因。而Ziehr等人在一组COVID-19患者中描述了与Berlin ARDS的定义一致的低顺应性表现。值得注意的是,机械通气患者的COVID-19最严重,因此可能呼吸系统顺应性最低。呼吸困难本身可能促使机械通气,机械通气可能就代表了COVID-19的低顺应性。随着进一步研究的报道,对COVID-19呼吸力学的了解将不断深入。
快速恶化
低氧血症驱动的呼吸急促、过强呼吸和氧合改变可预测疾病严重程度和/或宿主反应和/或次优管理诱导的临床恶化。随着疾病的进展,跨肺压升高,越来越实变的气腔不容易充气。在较高的肺容积下,容积损失按比例更大。这种肺容量的减少降低了肺总顺应性并增加了呼吸功。如肺炎球菌肺炎中所见,有证据表明,SARS-CoV-2肺炎中剩余通气肺的动态顺应性降低,这最可能是由于表面活性物质活性降低,这就进一步增加了呼吸功。由于血管内血栓引起的血流量减少,生理性无效腔也增加。重要的是,COVID-19患者经历的焦虑也会影响对呼吸中枢的皮层反馈。因此,随着疾病进展,呼吸困难越来越明显。
对管理的思考
当COVID-19患者因低氧血症入院时,病毒复制正顺利进行,除给予抗病毒药物外,纠正V/Q失调和减少细胞因子风暴仍是主要的治疗目标。关于血流,避免微血栓和持续的纤维蛋白沉积是治疗策略之一。在所有COVID-19患者中预防血栓似乎是明智的,尤其是入院时D二聚体较高的患者。Moore等人最近建议使用组织型纤溶酶原激活物(tPA)治疗 COVID-19的ARDS。此外,为预防大血栓和微血栓,使用抗炎药物(如抗IL6R托珠单抗或sarilumab,或抗IL6抗体司妥昔单抗或补体抑制策略)来处理全身血栓前并发症是另一种潜在的方法,目前几项研究正在验证这一假设。改善低氧性肺血管收缩可能是改善肺局部灌注和通气匹配的另一靶点。炎症介质过度释放,干扰了肺毛细血管中一氧化氮(NO)、内皮素和前列腺素间的平衡,但吸入NO始终未能改善ARDS的死亡率 。RAS调节(例如血管紧张素受体阻滞剂、重组可溶性ACE2和缓激肽系统抑制)可能在恢复肺灌注调节中具有潜在作用,研究正在进行中。
关于通气,氧疗才是改善氧合的第一步。在难治性低氧血症性呼吸衰竭(分流率增加)的患者中,相比无创通气,及时(而非过早)插管和有创通气支持,在提高跨肺压、复张塌陷的肺泡、改善氧合、减少氧债、避免P-SILI和为肺部提供更好的愈合机会方面可能更优。至于重症病例,大多数患者符合ARDS的Berlin标准,其中肺保护通气、俯卧位通气、有效镇静镇痛以及高呼气末正压(PEEP)是关键。PEEP非常容易造成COVID-19患者肺损伤,适度耐受允许性高碳酸血症可最大限度地减少呼吸机诱导的肺损伤(VILI)。由于俯卧位复张背侧肺区并转移血流至足侧肺区,因此在COVID-19早期和相对较长的病程中可能特别有意义。尽管还需进一步的研究来评价对疾病严重程度和死亡率的影响,但某些作者证实,清醒俯卧可改善COVID-19的氧合。
结论
严重低氧血症性呼吸衰竭和临床“快乐”患者间的显著脱节很常见,这应该提醒医生和医护人员不仅依靠患者的表观健康,还要密切监测呼吸频率、过度通气体征、血氧饱和度和低氧血症/低碳酸血症的有创测量。由于氧合血红蛋白解离曲线左移,解释脉搏血氧饱和度应谨慎。动脉低氧血症是由肺内分流、缺氧性肺血管收缩失调、肺弥散障碍和血管内微血栓导致。疾病的前几天,肺力学完好,无气道阻力增加或无效腔通气。因此,呼吸中枢不会有到呼吸不适的感觉。然而,可能出现突然和快速的呼吸失代偿,呼吸急促和呼吸过度可能是COVID-19患者即将发生呼吸衰竭最重要临床警示体征。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








太有收获了
82