
Nat Commun:王建新/胡斌教授团队建立基于动态边界调整算法的转座子元件检测和注释方法
2024-07-07 测序中国 测序中国 发表于上海

基于基因组组装数据,提出了快速准确的转座子识别和注释算法,并开发了相应的软件HiTE。
转座子(TEs)占据了大多数真核生物的重复区域,并已知对基因组进化和种内基因组多样性有显著影响。研究发现,TEs通过中断或调控关键基因在人体疾病和作物育种中发挥重要作用。然而,识别完整的TEs具有挑战性,因为存在多种复杂情况,包括但不限于:(i) TEs的降解速率不同,可能导致结构信号的丧失;(ii) 由于随机的删除、插入和嵌套TE,TE序列呈现复杂模式;(iii) 难以确定高度碎片化TE实例的真实末端;(iv) 丰富的碎片化TE阻碍构建全长TE模型;(v) 无关TE之间的区域同源性对其识别和分类造成干扰;(vi) 误将高拷贝数的片段重复或串联重复识别为潜在TE实例的风险。近年来,基因组组装技术的进步极大改善了对转座元件进行全面注释的前景。然而,现有基于基因组组装的TE注释方法由于缺乏准确性和鲁棒性,仍需要大量的人工编辑。此外,即使是对已广泛研究的物种,目前可用的黄金标准TE数据库也不够全面,迫切需要一种自动化的TE检测方法来补充现有的数据库。
2024年7月2日,中南大学计算机学院王建新教授和北京理工大学医学技术学院胡斌教授等在Nature Communications上在线发表题为“HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable elements detection and annotation”的研究论文,基于基因组组装数据,提出了快速准确的转座子识别和注释算法,并开发了相应的软件HiTE。中南大学计算机学院胡康、倪鹏为论文共同第一作者,中南大学为第一署名单位,该研究受国家重点研发计划、国家自然科学基金原创项目、国家自然科学基金重点项目、湘江实验室揭榜挂帅项目等多个项目支持。

文章发表在Nature Communications
HiTE采用多种检测方法来注释基因组上不同类型转座子,包括LTR、non-LTR、TIR以及Helitron转座子。HiTE首先利用转座子的重复特性在基因组上从头检测 “粗”边界的候选转座子元件(图1a-d),然后使用转座子的结构特征在候选转座子元件中进一步确定转座子元件的“细”边界(图1e),接着实现基于动态边界调整方法对转座子拷贝的多序列比对结果进行动态识别,进一步确定转座子的精确边界并过滤绝大多数假阳性序列(图1f)。此外,HiTE分别利用多种结构特征(图1h)和同源性(图1i)识别基因组上的LTR和non-LTR元件。最后汇合多种转座子序列,去除其中的嵌合和冗余,生成高质量转座子数据库(图1g)。
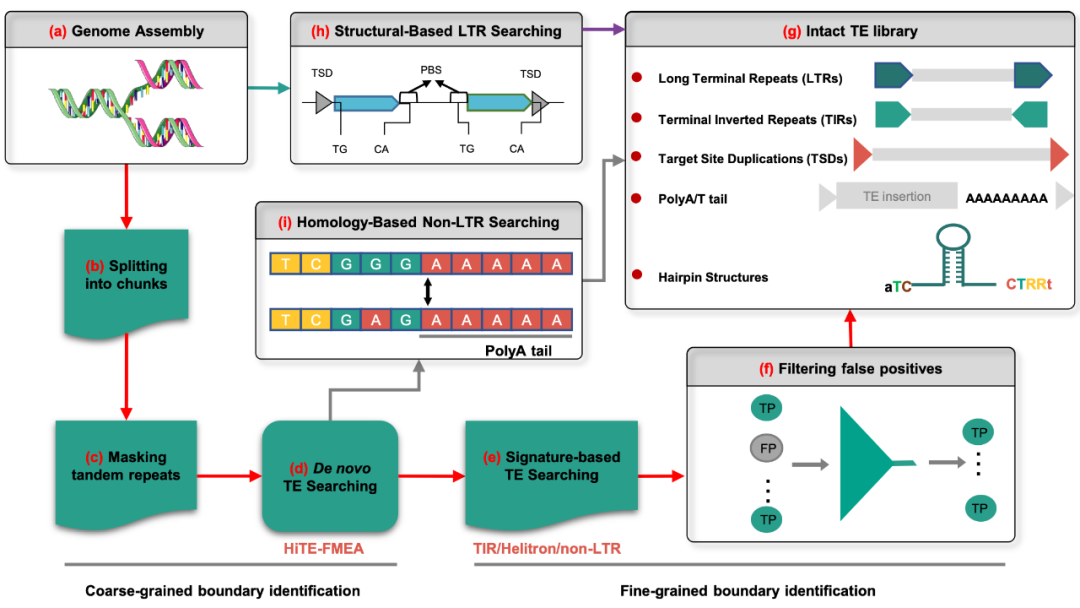
图1. HiTE注释流程图
研究团队首先对已有方法RepeatModeler2和EDTA的评价方法进行了改进,并提出了一种更为精确的评价方法,重新对转座子注释的真阳性和假阳性进行定义,减轻了碎片化序列对结果的影响,更适合评估高质量的全长转座子库(图2)。此外,由于生成转座子一致性序列的差异阈值并未统一,目前存在80%、90%、95%和99%等多种不同的阈值,新的评价方式可以在不同阈值下评估转座子的识别和注释性能。
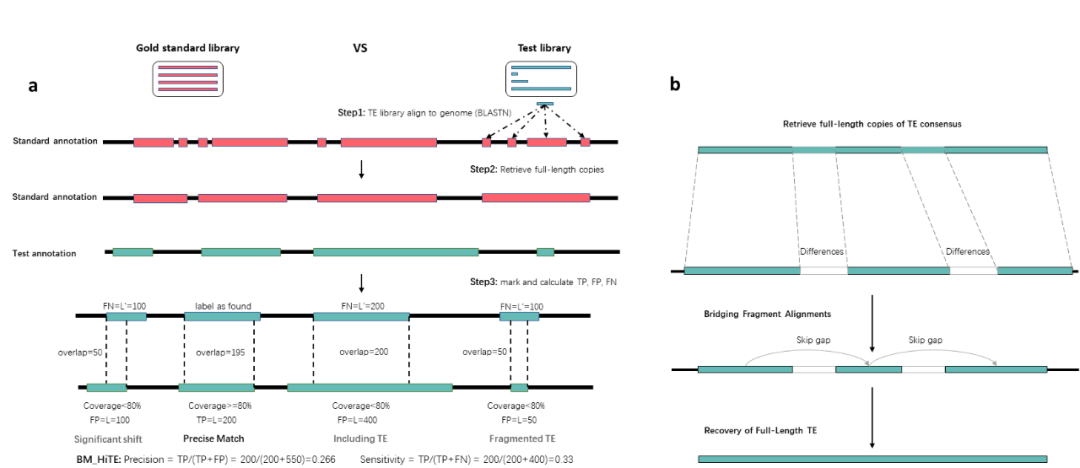
图2. HiTE评价方法示意图
研究团队接着使用包含RepeatModeler2、EDTA和HiTE在内的三种评价方法对HiTE的总体性能进行了测试,评估其对于全长转座子的识别和注释能力。如图3a所示,在95%阈值下,HiTE识别了远超已有工具的全长TE数量和F1值,例如在水稻中识别了1,078个全长的TE序列,并获得了93.56%的F1值,而已有最先进方法EDTA和RepeatModeler2分别只识别了506和378个全长序列以及87.34%和54.82%的F1值。在其他物种和更严格的99%阈值(图3b)下获得了相似的结论,说明HiTE具有可靠的泛化能力。
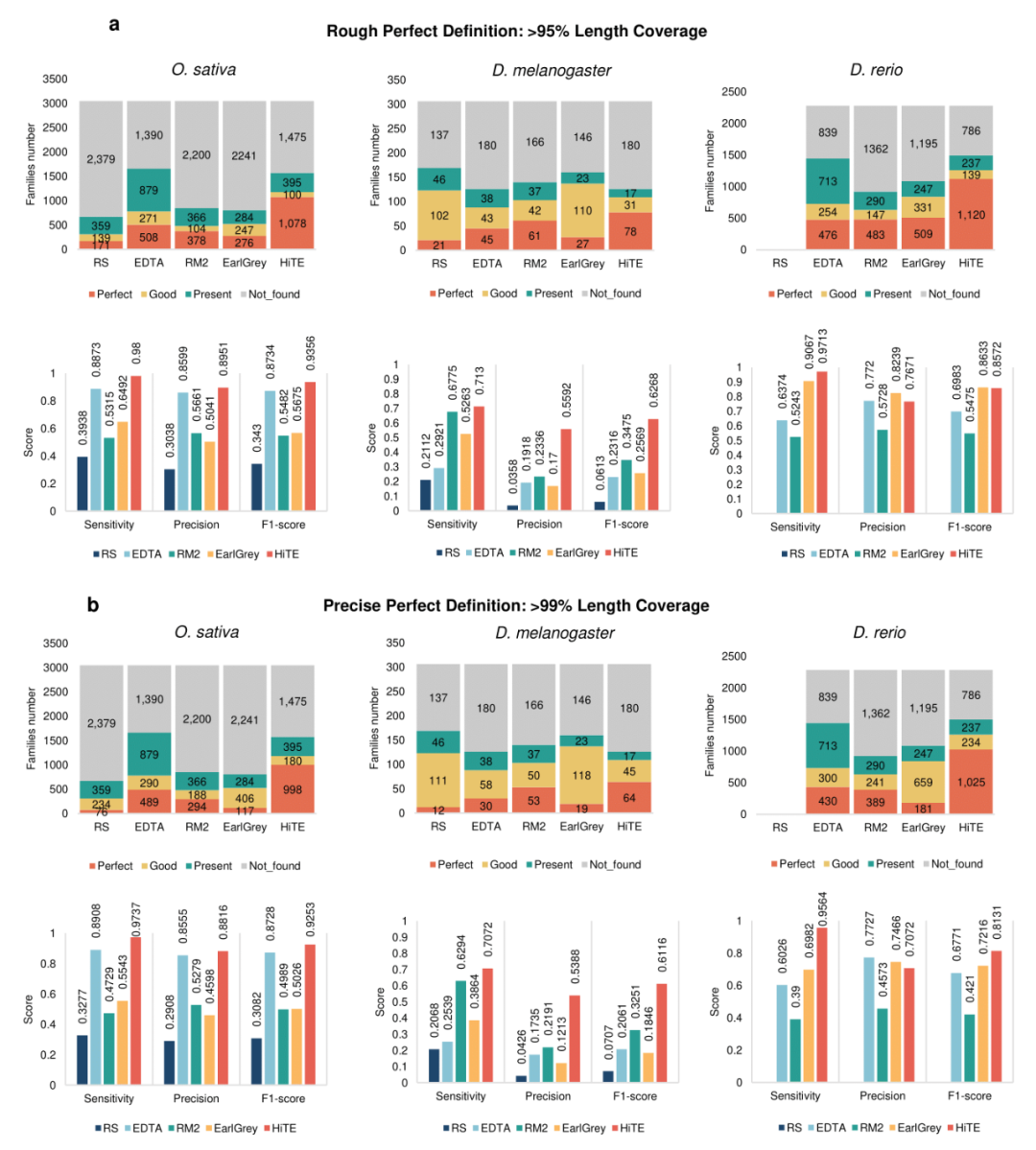
图3. HiTE识别转座子性能评估
研究团队进一步对HiTE的多个不同模块进行了评估。在水稻中,使用HiTE中提出的de novo检测“粗”边界的候选转座子方法FMEA(图4a),获得了远超已有方法的全长TE数量(图4c),然而由于并未过滤结果中大量的假阳性序列,导致FMEA获得较低的精度(图4b)。为了解决这一问题,研究团队实现了动态边界调整的过滤方法,能够根据转座子拷贝的多序列比对结果对边界进行动态识别和调整(图5a),并过滤掉大量的假阳性序列。实验结果表明,过滤后的HiTE获得了更多的全长TE数量(图5c),并且在几乎未降低敏感性的情况下,大幅度地提升了识别精度(图5b)。此外,研究团队比较了不同工具在特定类型转座子如TIR和Helitron识别上的性能,HiTE仍获得了最高的性能(图5d和e)。
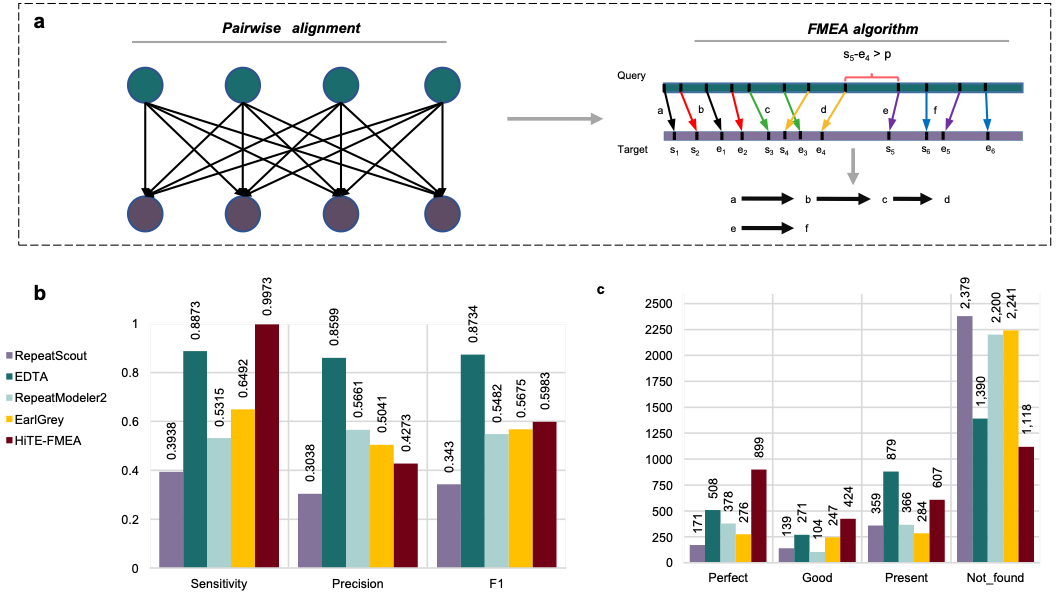
图4.基于水稻基因组的HiTE de novo方法性能评估
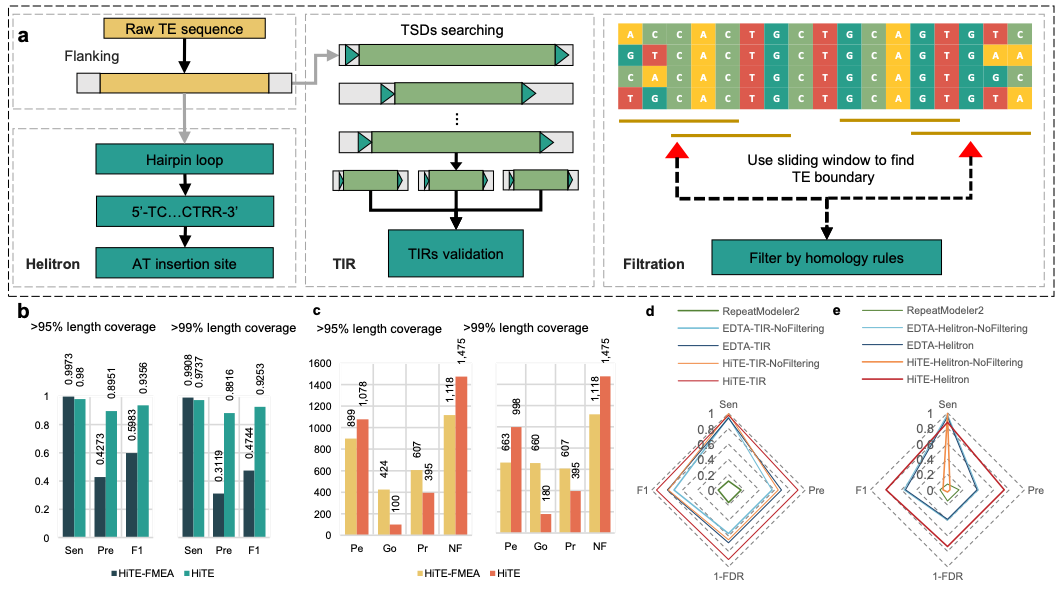
图5. 基于动态边界调整算法的HiTE过滤性能评估
HiTE可以发现具有保守结构特征和多个拷贝的新型转座子。Ghd2基因(LOC_Os02g49880)在调控水稻的重要农艺性状方面起着关键作用,如植株高度、籽粒数量和抽穗日期。已有研究表明在Ghd2基因的3’端非编码区存在一个微型反向重复转座元件sMITE,起到了转录抑制剂的功能,研究团队观察并验证了这一点。此外,研究团队还检测到了一个新的TIR元件(Novel_TIR_Ghd2)紧邻该sMITE,并呈现出截断状态(图6a)。通过人工验证,确认其是一个真正的转座子,具有9-10 bp的TSDs和224 bp的终端反向重复,属于Mutator超家族(图6e)。RepeatModeler2同样识别了该转座子,但其识别边界并不完整,无法在没有后续人工编辑的情况下识别TSD等结构特征(图6c)。考虑到sMITE经常插入其他转座子(图6d),研究团队推测在远古水稻中,Ghd2基因最初是由Novel_TIR_Ghd2调控的。然而,sMITE插入Novel_TIR_Ghd2导致了其原始调控角色的改变,最终使其逐渐退化,这可能在水稻的进化过程中起到了关键作用。为了证实该推测,研究团队比较了sMITE和Novel_TIR_Ghd2的拷贝数和终端反向重复序列的一致性(图6b)。结果显示,sMITE具有429个拷贝和较高的终端一致性,与仅有8个拷贝的Novel_TIR_Ghd2相比,其活性更高。考虑到水稻的突变率为1.3×10-8每位点/每年,据估计,sMITE和Novel_TIR_Ghd2的插入时间分别为526和948万年前。
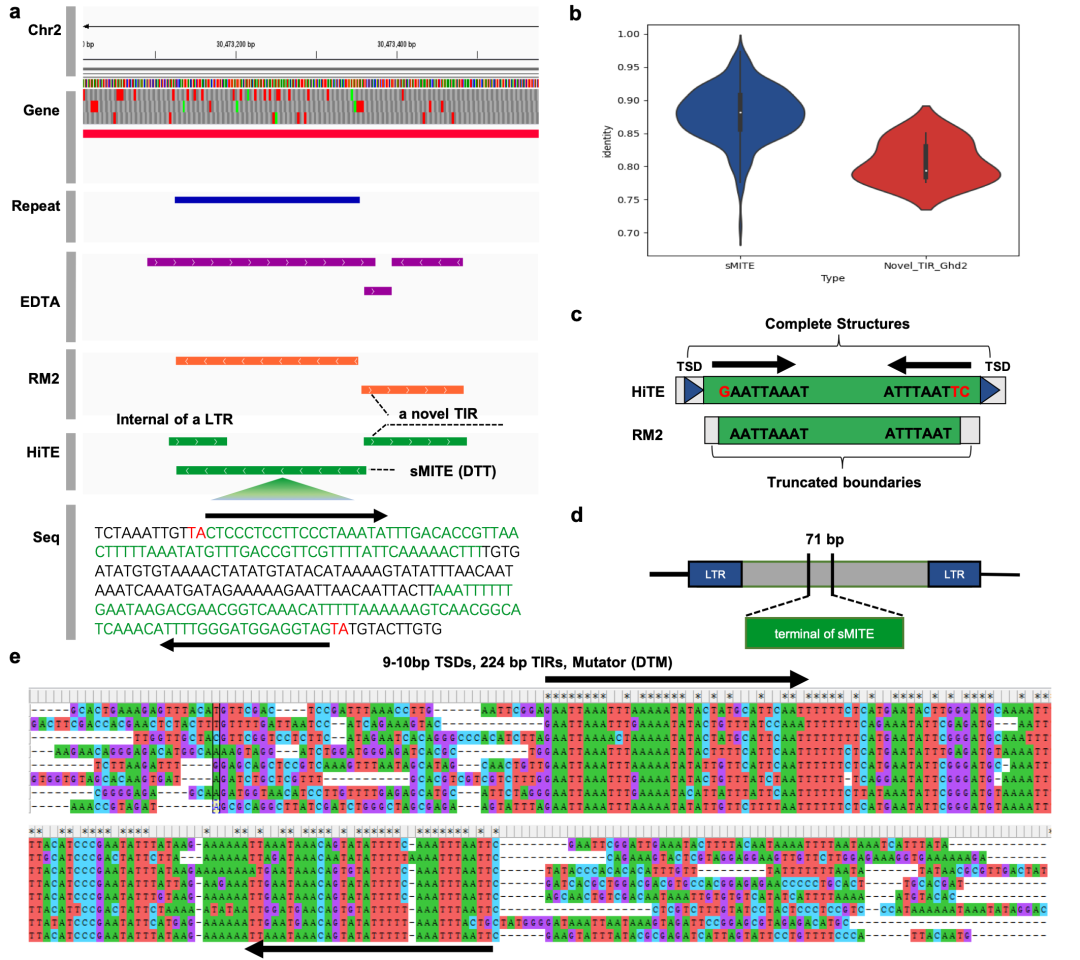
图6. HiTE发现了水稻中MITE与关键基因Ghd2之间一个未曾揭示的现象
综上所述,研究团队提出了基于动态边界调整算法的转座子检测和注释方法HiTE。在不同数据集上的测试结果表明,HiTE相比已有工具能够获得更多的全长转座子序列,且具有更高的精度和敏感性,同时能够发现尚未被识别的转座子序列,有助于基因组变异研究分析,在人类疾病和作物育种等领域具有较高的应用潜力。此外,随着测序技术的发展,更多新物种组装数据开始涌现,HiTE相比已有方法能更快获得高质量的转座子数据库,加快新物种的基因组注释流程。
文章第一作者:
胡康,中南大学计算机学院博士。主要从事转座子的识别和注释,开发了HiTE、NeuralTE等转座子识别和注释工具,在Nature Communications、Bioinformatics等期刊发表多篇文章。
倪鹏,中南大学计算机学院讲师。主要从事三代测序数据分析和疾病关联模式挖掘等研究,开发了甲基化检测工具DeepSignal、DeepSignal-plant、ccsmeth等,在Nature Communications、Bioinformatics等期刊发表文章10余篇。
论文原文:
Hu, K., Ni, P., Xu, M. et al. HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable element detection and annotation. Nat Commun 15, 5573 (2024). https://doi.org/10.1038/s41467-024-49912-8
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#转座子# #基因组组装# #HiTE#
20