
论文解读│陈华群教授团队发现靶向TMEM16A可防止恶性黑色素瘤BRAFi药物耐药性产生
2024-08-29 Genes and Diseases Genes and Diseases 发表于上海

该研究为解决BRAFi类药物耐药性问题提供了全新思路,同时也提出一个新的治疗恶性黑色素瘤的多效应的单分子靶点。
BRAF(V600E)突变是恶性黑色素瘤的一种常见突变。业已发现,MEK/ERK信号通路的持续激活是导致这一肿瘤生长的关键。根据这一特征,人们设计成功了多种BRAF 抑制(BRAFi)药物和MEK抑制(MEKi)药物。自2011年美国和欧盟批准使用以来,这些靶向药物—BRAFi单药如维莫非尼(vemurafenib, VMF)、达帕菲尼(dabrafenib),及BRAFi联合MEKi药物如曲美替尼(trametinib)的使用对于恶性黑色素瘤的治疗取得了很大的成功,病人的生存率得到显著提高。遗憾的是,原发和获得性耐药严重影响了这些药物的应用。如何防止耐药性、是否有新的药物靶点是恶性黑色素瘤药物开发的难题。
南京师范大学陈华群课题组在本刊发表了题为“Targeting TMEM16A/ANO1 inhibits the progression of BRAF mutant (V600E) melanoma through the MEK/ERK and AKT signaling pathways”的研究快讯。作者团队通过生物信息学研究发现,恶性黑色素瘤病人肿瘤组织中TMEM16A(一种钙依赖的氯通道蛋白)表达水平升高,并与病人预后相关(图1A–C)。进一步发现,携带BRAF(V600E)突变的人恶性黑色素瘤细胞株A375中TMEM16A表达水平也呈现异常升高(图1D)。利用抑制剂和shRNA慢病毒介导的TMEM16A敲降研究发现,抑制TMEM16A显著抑制A375细胞的增殖(图1E-H)。裸鼠体内研究同样发现,敲降TMEM16A的A375移植瘤的生长被显著抑制(图1J、K)。体内外研究均发现,TMEM16A敲降的A375细胞中,不但MEK/ERK的激活被抑制, AKT的激活也被抑制(图1I、 L、M)。该研究还发现,在恶性黑色素瘤细胞中,敲降TMEM16A对于VMF诱发的MEK/ERK激活水平降低具有加强作用,表现为明显的协同作用;而VMF处理则诱发了明显的AKT激活,敲降TMEM16A可显著降低AKT的激活水平(图1O、P)。鉴于BRAFi药物耐药的重要原因是药物诱导的AKT激活,该结果强烈提示,抑制TMEM16A可以抑制黑色素瘤对于BRAFi药物的耐药性。
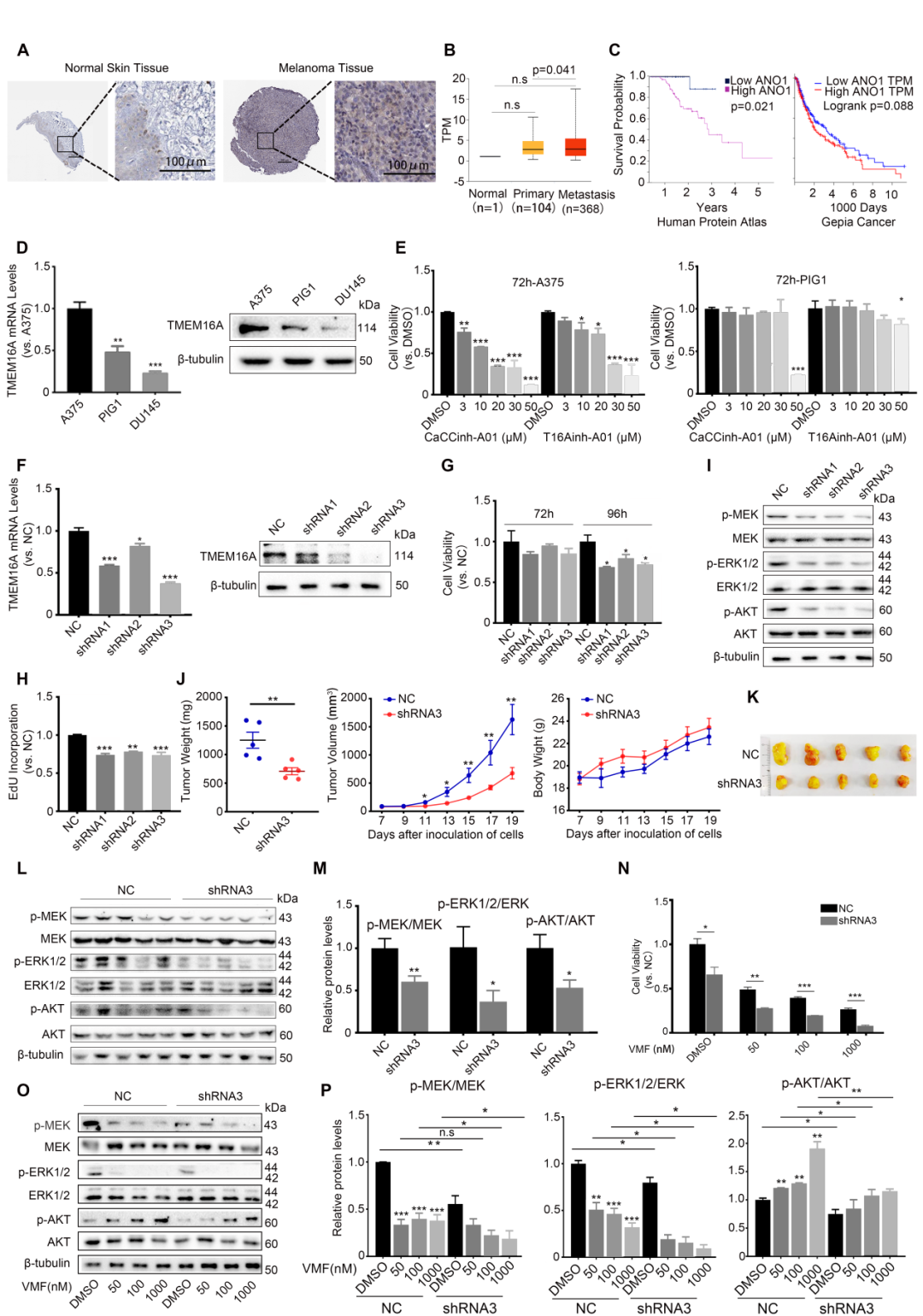
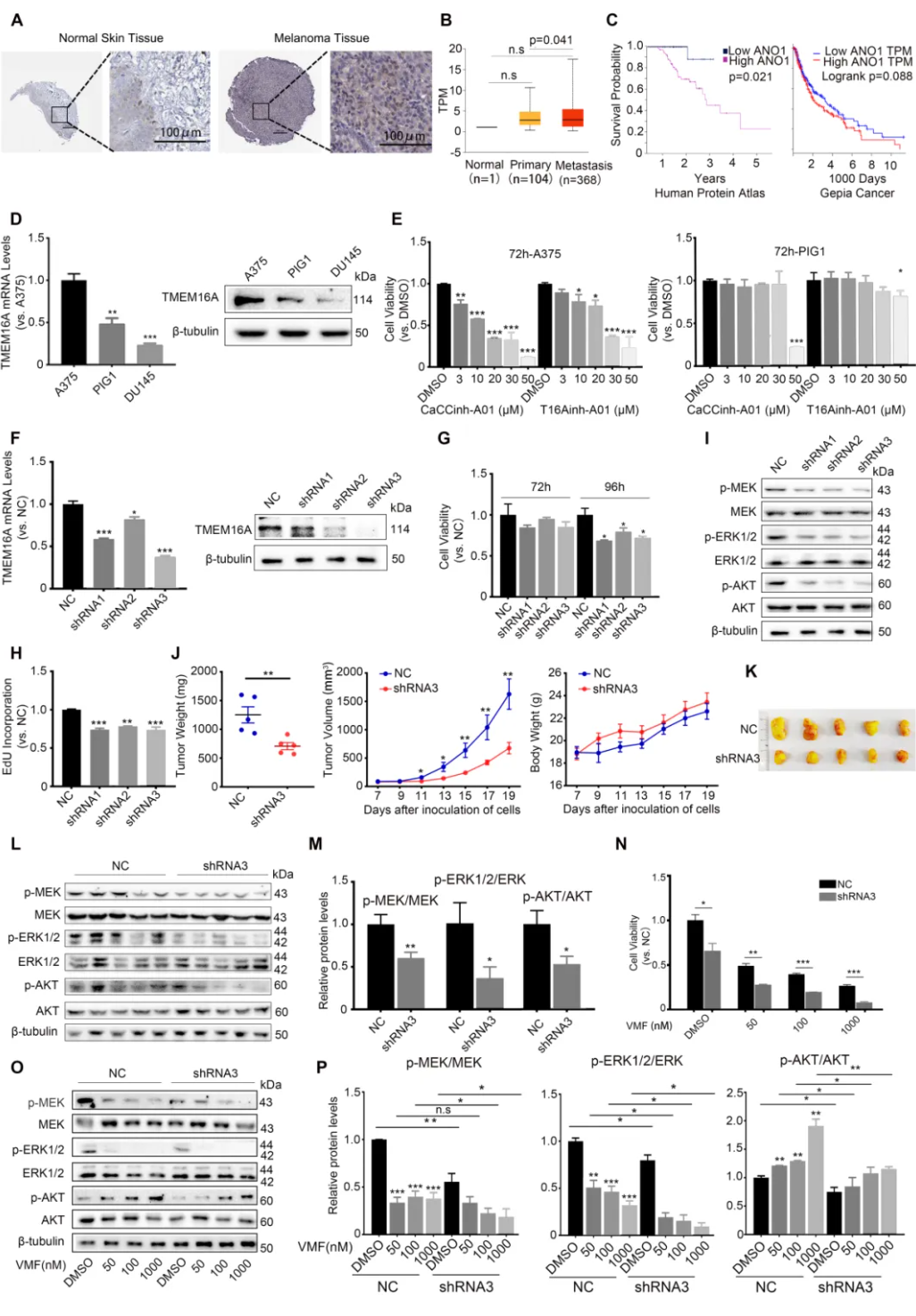 图1 靶向TMEM16A/ANO1抑制恶性黑色素瘤的发展和VMF耐药性。
图1 靶向TMEM16A/ANO1抑制恶性黑色素瘤的发展和VMF耐药性。
综上所述,研究者认为:TMEM16A抑制药物不但可协同加强BRAFi药物的MEK/ERK抑制效应和抑制恶性黑色素瘤生长的效应,还可通过抑制AKT活性减轻BRAFi药物耐药性的产生。该研究为解决BRAFi类药物耐药性问题提供了全新思路,同时也提出一个新的治疗恶性黑色素瘤的多效应的单分子靶点。
文章来源
免费全文下载链接:
https://www.sciencedirect.com/science/article/pii/S2352304223004361
引用这篇文章:
Shi M, Wang J, Huang X, et al. Targeting TMEM16A/ANO1 inhibits the progression of BRAF mutant (V600E) melanoma through the MEK/ERK and AKT signaling pathways. Genes Dis. 2024;11(6):101153.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言














#黑色素瘤# #TMEM16A#
81