Genome Biol:scRNA-seq辅助分析新方法scGWAS,揭示性状-细胞类型关联图谱
2022-11-18 测序中国 测序中国

单细胞RNA测序(scRNA-seq)技术可以定量测量单个细胞的基因表达,能够检测细胞类型特异性的转录组特征,为解码复杂疾病中由遗传介导的细胞类型特异性提供了独特的机会。
全基因组关联研究(GWAS)中报告的近90%疾病易感性位点都位于非编码区,且被预测可能发挥调节作用。遗传调控具有高度组织和细胞类型特异性。一种细胞类型通常与多种复杂的性状相关联,性状和细胞类型关联也对疾病并发症和动态进展有影响。因此,识别性状和细胞类型之间的遗传介导关联对于理解遗传变异的功能影响和潜在的疾病机制至关重要。
单细胞RNA测序(scRNA-seq)技术可以定量测量单个细胞的基因表达,能够检测细胞类型特异性的转录组特征,为解码复杂疾病中由遗传介导的细胞类型特异性提供了独特的机会。将scRNA-seq数据整合到GWAS分析中有助于人们发现人类复杂性状的细胞类型特异性。
近日,美国德克萨斯大学休斯顿健康科学中心的Zhongming Zhao教授团队在Genome Biology上发表了题为“scGWAS:landscape of trait-cell type associations by integrating single-cell transcriptomics-wide and genome-wide association studies”的文章。研究团队提出了一种新的scRNA-seq辅助分析方法“scGWAS”,通过利用人类基因组中的各种基因关联来研究特定细胞类型背景下遗传变异的转录变化。scGWAS能够有效利用scRNA-seq数据来推断疾病相关基因表现的细胞类型,识别细胞类型和性状之间的遗传介导关联,揭示不同的疾病特异性激活过程。 文章发表在Genome Biology
文章发表在Genome Biology
研究团队从九个主要人体组织(血液、脑、蜕膜、食道、心脏、肝脏、肺、胰腺和脾脏)收集了18个scRNA-seq数据集,还收集了包含40个具有代表性复杂性状的GWAS汇总统计数据集。研究人员使用deTS和GTEx等大体积转录组数据对上述性状的组织特异性进行了初步调查(图1),鉴定出了许多特征与组织的关联,其中大多数与生物学预期一致。值得注意的是,虽然一些关联缺乏明显的生物学联系,但有四种组织(全血、肺、脾和小肠末端回肠)始终与免疫调节中常见的几种疾病相关。
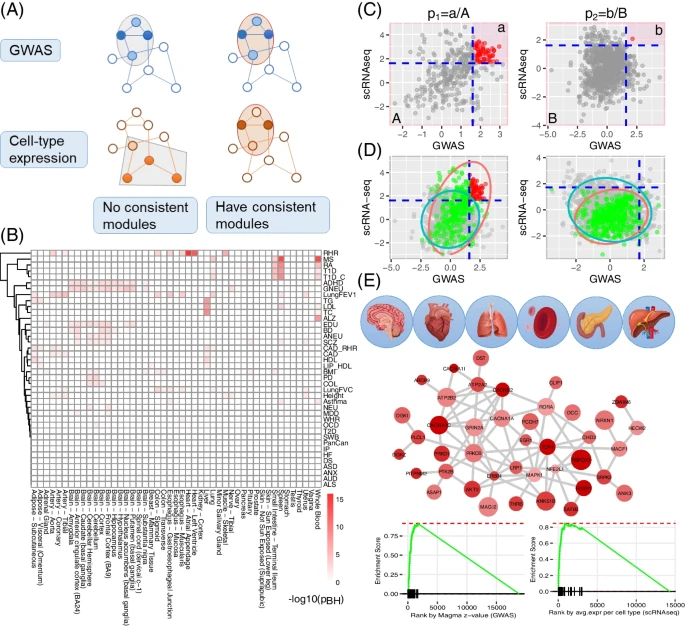 图1. 解码性状相关组织和细胞类型的分析框架。
图1. 解码性状相关组织和细胞类型的分析框架。
scGWAS的原理如图1A所示。scGWAS有两个目标:一是确定GWAS暗示的基因是否在特定的细胞类型中被一致激活;二是识别遗传关联信号和细胞类型表达信号都显著富集的基因模块。在scGWAS中,研究团队设计了几个重要的步骤来验证结果的准确性。首先,提出了一种新的程序来规范GWAS数据和scRNA-seq数据,以便对其进行集成(图2);其次,开发了一种序列模块扩展-反向检测(MEBE)算法,以GWAS和细胞表达数据加权的异质信息来构建基因模块。
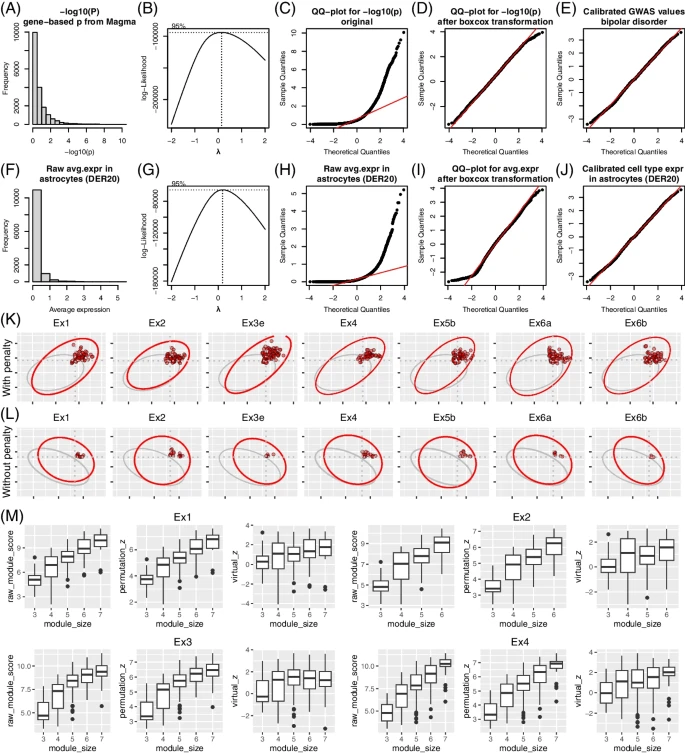 图2. scGWAS方法概述。
图2. scGWAS方法概述。
研究团队通过18个单细胞panel(single-cell panel)将scGWAS应用在40个GWAS数据集中,共报告了437种组织细胞类型,最终确定了2,533个性状-细胞类型的关联。结果显示,在18个panel中,小胶质细胞、两个兴奋性神经元(Ex2和Ex8)和星形细胞等四个大脑panel显示的关联数量最多。在所有性状中,冠心病静息心率(CAD_RHR)的关联数量最多,其次是抑郁症状(DS)、阿尔茨海默病(ALZ)、冠心病(CAD)、1型糖尿病(T1D)。身高、心力衰竭、肺功能和内化问题等四个性状的关联数量最少。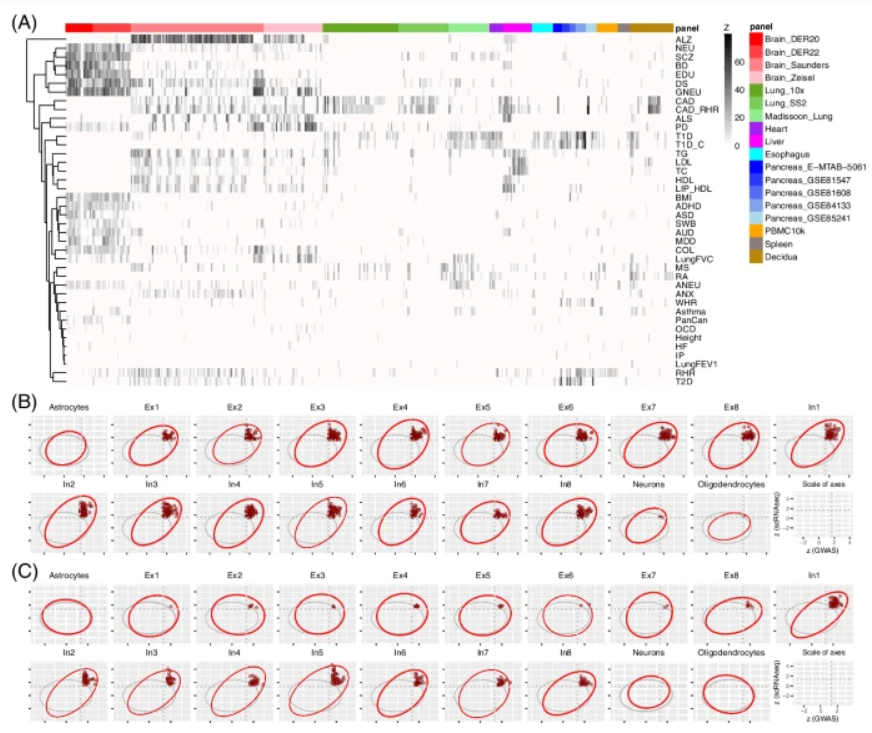 图3. scGWAS结果图示。
图3. scGWAS结果图示。
接下来,研究团队进行了一系列富集分析以评估由scGWAS识别的模块基因,使用基于ExAC数据集中带有pLI注释的基因、ClinVar数据集和OMIM基因对其进行验证。结果显示,与多数GWAS促进的基因和表达促进的基因相比,scGWAS鉴定出的模块基因在大多数组织和三个重要功能基因组中的表现更优异。
此外,研究团队还使用GWAS数据构建了每个性状的子网络(GWAS_only)以实现相同的模块搜索和虚拟搜索过程(如图4A所示),并将其与scGWAS搜索结果进行对比。在功能上,由scGWAS鉴定的模块基因往往比仅使用GWAS确定的模块基因更重要。
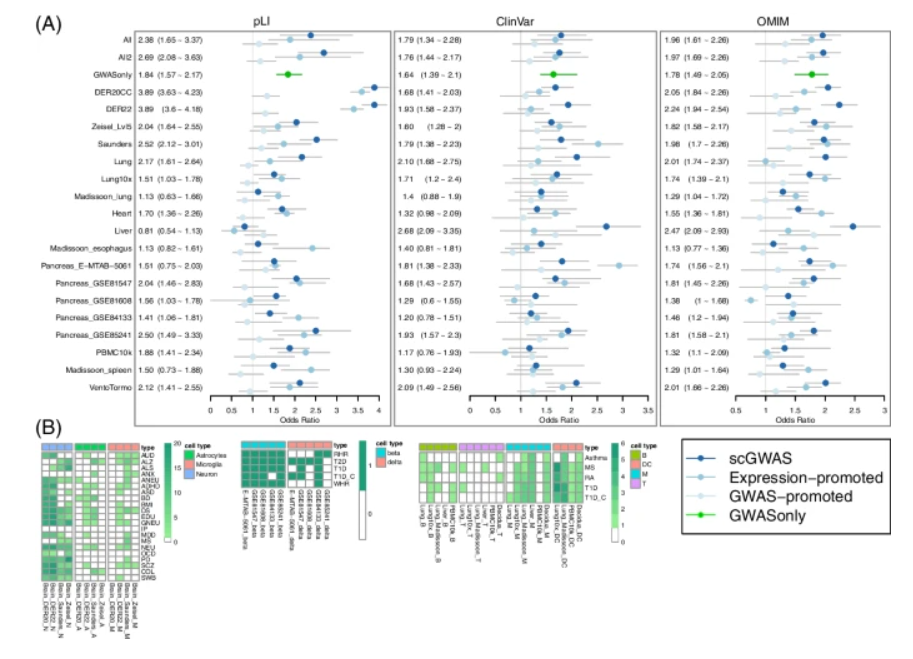 图4. 独立验证scGWAS结果。
图4. 独立验证scGWAS结果。
研究团队选择了由多个panel报告的细胞类型来进行性状-细胞关联的panel评估,其主要是来自大脑、胰腺和免疫系统的细胞。跨panel比较表明,scGWAS发现的关联是可靠且可复制的。
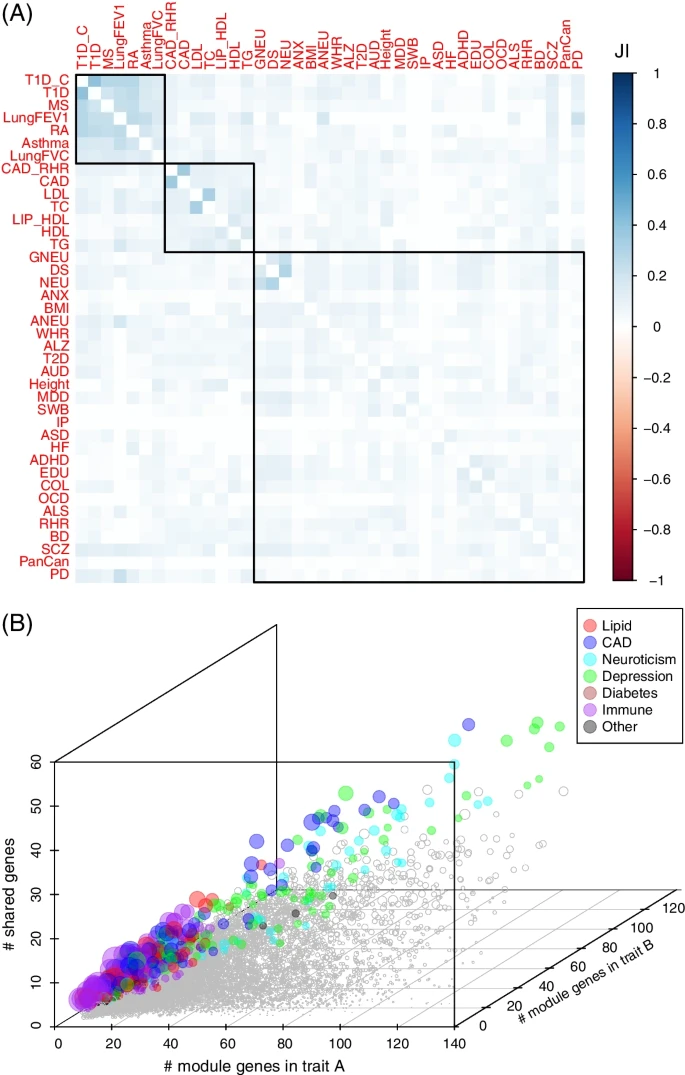 图5. 基于模块基因的性状间相关性研究。
图5. 基于模块基因的性状间相关性研究。
考虑到多个性状或疾病可能与相同的细胞类型有关,研究团队分析了研究性状中的共享基因。首先收集了每种细胞类型的相关性状,并根据模块基因计算了Jaccard指数(JI),确定了三个主要性状组:免疫(哮喘,T1D,T1D_C,MS和RA),脂质(TG,HDL,LIP_HDL和TC)和脑部疾病(GNEU,DS,NEU,ALS等)。通过检查高度重叠基因的性状对,发现同一组中的性状通常比与非相关组的性状具有更多的共享基因。上述结果表明,共享遗传成分的性状在同一细胞类型中往往具有更多的模块基因。
此外,研究团队还发现了许多HLA基因(如HLA- dma、HLA- dqa2、HLA- dqb2、HLA- drb5和HLA- f)以及其他基因(如TNF)介导的性状和免疫细胞关联(图6)。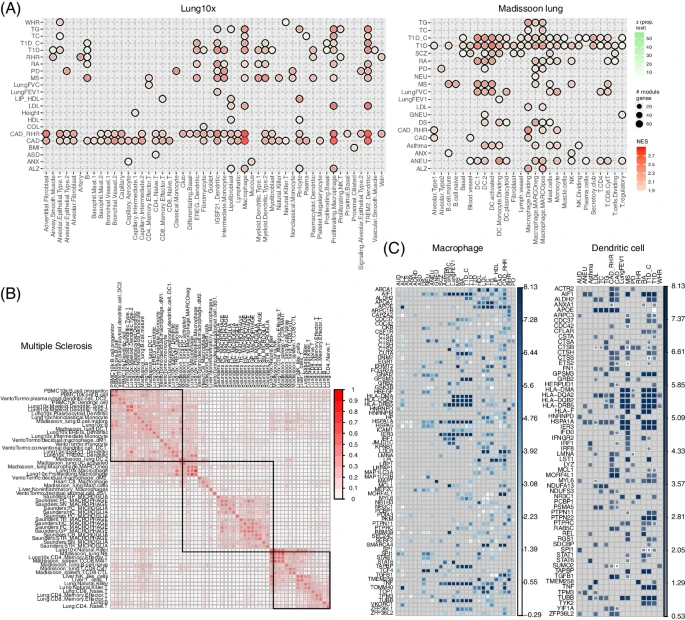 图6. 利用肺panel进行特征细胞类型关联。
图6. 利用肺panel进行特征细胞类型关联。
综上所述,研究团队开发了一种新方法——scGWAS,并将其应用于40个涵盖了各种复杂人类特征的GWAS数据集中。分析了许多已被充分研究的性状组织-细胞类型关联,并进一步揭示了先前未报道的新关联,为未解决的特征组织关联提供了见解。总而言之,该研究表明scGWAS是利用scRNA-seq数据在细胞类型水平上探索复杂疾病的有力工具。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







不错学习了。
0