
整理 | 毛冬蕾
主持人:EPS International株式会社事业开发部管理部事业开发课总监 王娣女士
嘉宾:EPS株式会社真实世界证据事业本部临床研究中心副主任森冈久代(Hisayo Morioka)女士
由药研社、EPS International 株式会社、日中医学交流中心(JCMCC)、研发客和药匠说举办的“临床研究在日本”系列讲座播出以来受到行业内的一致好评,每场在线人数不断增加。本期,来自EPS株式会社真实世界证据事业本部临床研究中心的副主任森冈久代女士介绍了日本的《临床研究法》和研究者发起的临床试验(IIT)。具体有三项内容,一是日本临床研究与注册临床试验间的差异;二是日本《临床研究法》的介绍;三是IIT与制药企业发起的临床试验的差异。
日本真实世界临床研究是什么
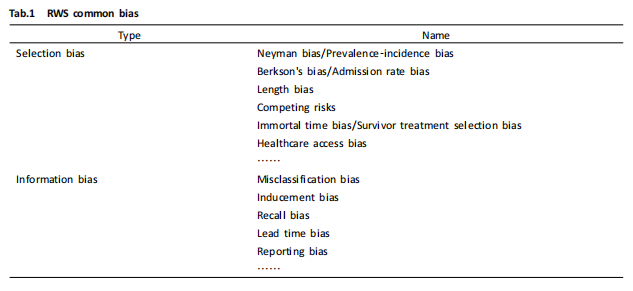
在日本,注册临床试验和注册临床试验以外的相关法律规定如下:
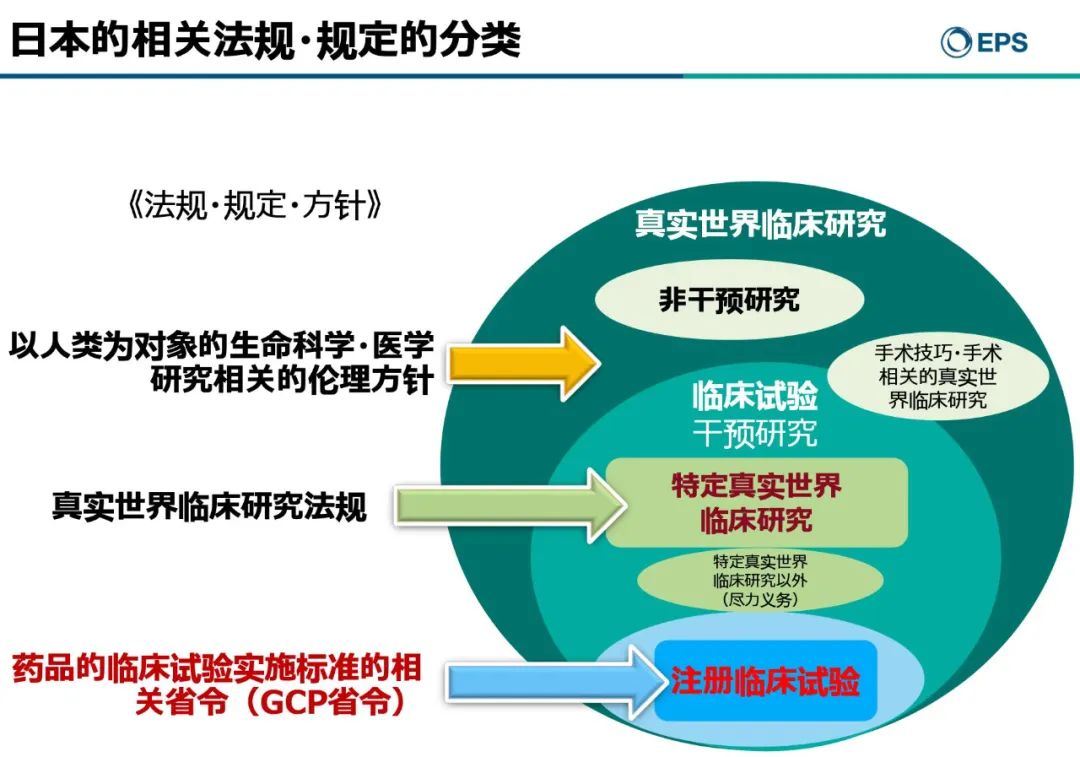
在上图中,森冈久代讲到,日本的真实世界研究涵盖的范围最广,包含所有以人类为对象进行的各种医学研究。通过分析在真实世界环境下收集与患者相关的研究数据,获得药品使用价值、获益或风险的临床证据,主要类型包括干预性临床试验及注册临床试验。
干预性临床试验是医疗人员开展的预防方法、诊断方法、诊疗方案、评价药物安全性和有效性为目的的试验;而注册临床试验则是申办方以上市为目的的药品和医疗器械研究。
需要指出,本次讲座中的真实世界临床研究是根据日本《药品和医疗器械法》,针对未获批药品/超适应症药品开展的临床研究,及由制药企业提供资金而开展的针对该企业药品的临床研究。
和我国一样,在日本,以上市注册为目的的临床试验要求和标准高于其它临床研究,并以《药品和医疗器械法》(相当于我国《药品管理法》)及日本GCP相关省令的法规执行。
《临床研究法》的规则和审查
森冈久代女士介绍说,日本《临床研究法》的出台有一定历史背景。早在上世纪90年代,日本发生了诺华的降压药缬沙坦事件,诺华与京都府立医科大学、东京慈惠会医科大学、滋贺医科大学、千叶大学及名古屋大学的研究者之间通过操纵试验数据等,令试验结果的可靠性受到质疑,与此同时,申办方和研究者之间的利益冲突已成为社会问题。诺华公司疑似涉嫌《药品和医疗器械法》违反规定的禁止夸大炒作,被刑事指控。

上述的事件,让日本厚生劳动省和产业界深度反思。除了以上市为目的的临床试验,所有以人类为研究和观察对象的临床试验都要设立法律加以监管。
日本厚生劳动省出台了《临床研究法》,并于2018年4月颁布执行。该法规定了真实世界研究和特定真实世界研究的开展流程,根据伦理委员会(CRB)的审查意见,切实符合开展临床试验的相关条件,并制定了与研究相关的经费信息公开制度等。确保以受试者权益为启始,国民对开展临床研究的信任,促进临床研究顺利开展,改善医疗卫生环境。
《临床研究法》第二条提到,临床研究是指药品或医疗器械等用于人体时,验证其有效性或安全性研究。森冈久代女士特别指出,《临床研究法》不适用于注册临床试验,上市后临床研究(PMS)和非干预研究。
《临床研究法》中对临床研究流程有详细规定。
在准备阶段:
首先,需要撰写试验方案,第二,需要通过临床研究审查委员会(CRB)审查,第三,获得开展试验医疗机构管理者的许可。第四,针对开展临床研究由厚生劳动省认可的临床研究审查委员会(CRB)审查临床研究计划,通过后才能向厚生劳动省提交临床研究计划,需要在指定的临床研究网站(jRCT: Japan Registry of Clinical Trials)办理相关备案手续。
在进行阶段:
首先,当发生严重不良事件时,如果无法排除是否是由于临床研究引起的不良反应,研究者需要报告给临床研究审查委员会(CRB)听取意见,同时需要报告给厚生劳动省。如果发生了非预期严重不良反应(SUSAR),还需要报告给PMDA。
其次,要根据药物风险程度,对临床研究方案进行监查。
第三,临床研究过程中出现方案变更时,与我国一样,按照风险的等级大小进行管理,如果是轻微的方案变更,在变更后10天内通知临床研究审查委员会。如果是重大变更时,在变更前需要告知厚生劳动省。在临床研究过程中,厚生劳动省有权对全过程进行监管,并提出改善要求,甚至可直接要求停止临床研究。
临床研究结束后10天内,研究发起人需要通知临床研究审查委员会,并向厚生劳动省提交备案文件。主要评价临床研究中的DB(Data Base,数据库)记录,而且需要在临床研究数据收集结束后的1年内完成撰写,不得有延迟。
《临床研究法》三个关键点
森冈久代女士特别指出,《临床研究法》主要包括三个关键点,一是临床研究审查委员会(CRB)审查、二是利益冲突管理、三是主要研究者职责。
首先,临床研究审查委员会需要厚生劳动省认定,由临床研究审查委员会进行集中的中心伦理审查,审查费用每个项目在3,000~8,000美元不等。
第二是利益冲突的管理COI (Conflict of Interest ) 。《临床研究法》规定,主要研究者、Sub-PI、统计分析负责人必须申报利益冲突,撰写相关利益冲突资料后提交临床研究审查委员会审议。在开展临床研究的医疗机构里,确认利益冲突的实际情况,并根据院长的要求,对申办方提供建议。
第三是主要研究者(PI)的职责,在IIT中主要研究者是申办方。其任务包括:
(1)撰写试验方案,提交给临床研究审查委员会和PMDA
(2)向临床研究审查委员会提交审查申请
(3)在j-RCT公开
(4)向临床研究审查委员会和厚生劳动省报告严重不良
生命医学伦理方针的简介
日本的《以人类为对象的生命科学和医学研究伦理方针》(以下简称《生命科学和医学研究伦理方针》)于 2021年6月执行;2022年4月部分修订,目的在于通过制定该方针,参与以人类为对象的生命科学和医学研究中各方应遵守的内容,确保人类尊严和人权得到保护,并促进研究合理进行。
以人类为对象的生命科学和医学研究的定义为:以人类为对象、以(a)或(b)为目的而开展的活动。具体而言如下:
(a)通过以下①②③④ 点,获得有助于保持和改善国民的健康或使患者恢复健康,以及提高生活质量的知识。
①伤病的原因
②病理的理解
③改善伤病的预防方法,或有效性验证
④改善医疗中的诊断方法和治疗方法,或通过性验证
(b)使用人体由来的样本・信息,获取有关人类遗传基因组及基因结构或功能,同时获取基因突变或发现相关知识。
《生命科学和医学研究伦理方针》的目的在于,当开展具有社会意义和学术意义的医学研究时,需根据医学研究领域特点,确保科学性和合理性;权衡受试者从医学研究中的获益与风险及其他不利因素;接受独立、公正的伦理委员会审查;事先需要向受试者充分说明,且受试者在自愿基础上获得知情同意;对社会弱势群体的受试者权益要给予特别考虑;妥善保管医学研究中的个人信息;最后需要确保研究的质量和透明度。
其主要关键点包括四点:
第一,规范化集中/中心审查
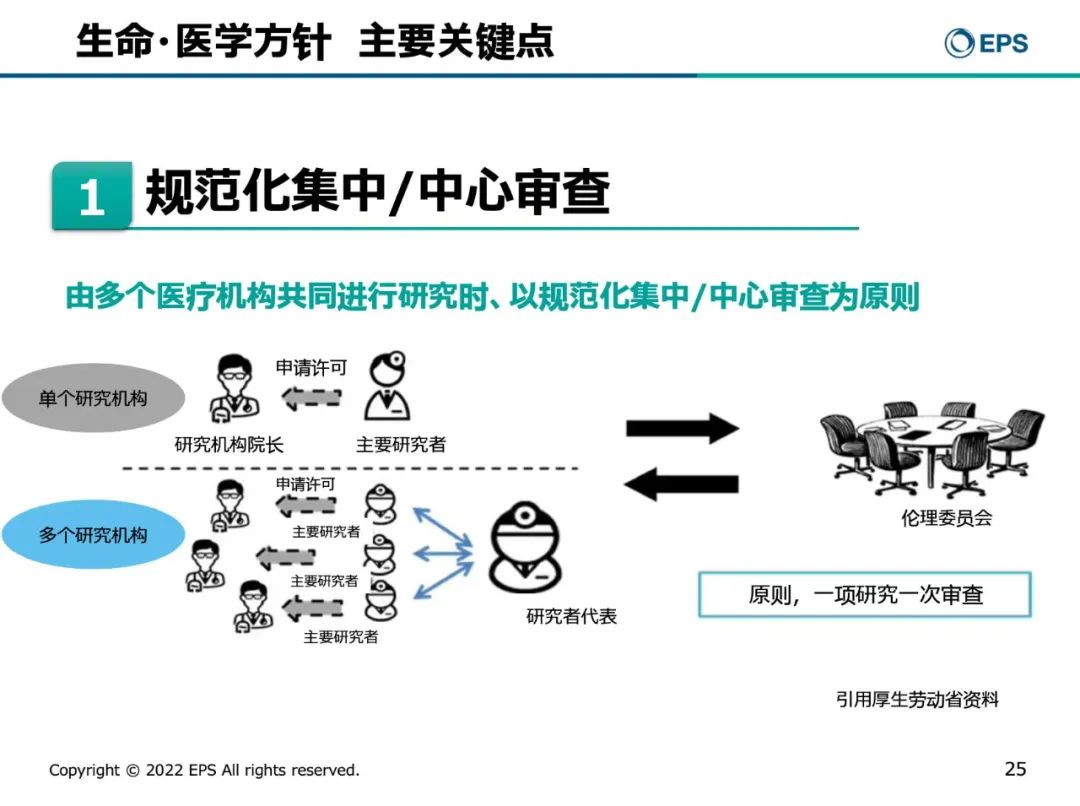 森冈久代女士强调,无论是单中心还是多中心,主要研究者要向研究机构的院长申请许可,提交伦理委员会上会申请,一项研究一次审查。
森冈久代女士强调,无论是单中心还是多中心,主要研究者要向研究机构的院长申请许可,提交伦理委员会上会申请,一项研究一次审查。
第二,设立新的“协助研究单位”
根据方案开展临床研究。研究单位以外的,为了该研究需要从研究对象获得新样本和信息,仅提供给研究单位。对无法获得知情同意的,协助研究单位为该研究获取新的人体样本和信息,研究单位在接收样本和信息时,必须获得研究人员等的知情同意。此外,协助研究的单位必须确认知情同意书是否通过适当的方式获得。SAE由PI把握,PI在向协助研究的单位获取与研究相关的样本和信息时, 如果研究对象发生严重不良反应,必须及时上报。
第三,电子知情同意
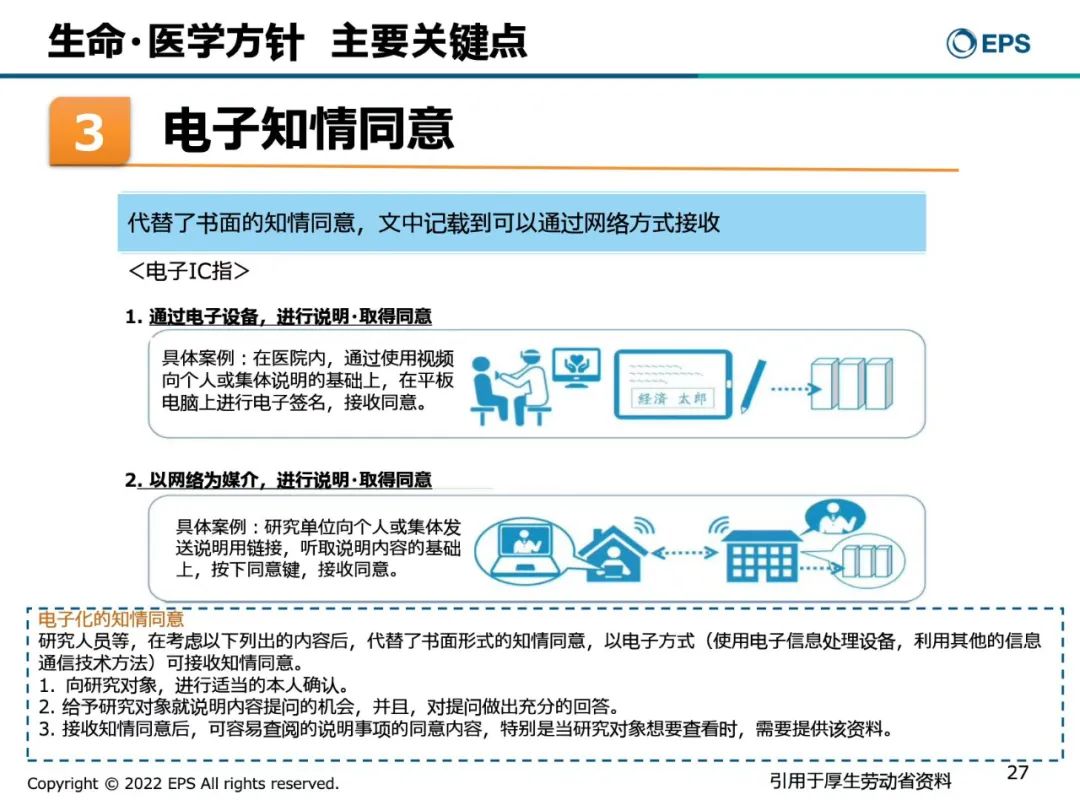
森冈久代女士指出,受新冠疫情影响,日本的各种临床试验中使用电子知情同意的情况十分普遍,已经取代了纸质知情同意,通过电子设备以网络为媒介,进行说明取得同意。最后在平板电脑上上进行电子签名,接收同意或者按下同意键,接收同意。
第四,伴随日本个人信息保护法的修订
日本的研究单位建立了“假名相关信息”(假名指非真实姓名,类似笔名等)。
最后,森冈久代女士提到,今后在日本,还需要进一步完善的课题:日本《临床研究法》流程繁琐,需要重新审查临床研究审查委员会的更新条件,从特定临床研究的适用范围除外的药物等;以人类为对象的生命科学・医学研究相关的伦理方针,伴随《个人信息保护法》的修订重新审查,以及重新审查知情同意手续。

日本的医学研究环境成就了IIT
日本各大国立医院大学附属医院的医生们十分热衷于参加和发起IIT。IIT在日本也是指由研究者发起的临床试验,与中国一样。IIT的发起的背景:第一,由于研究者是申办方,当一些患者数量少的疾病,或因药价低等盈利问题导致企业不愿意积极开发新药的药品・医疗器械,可以由研究者发起主导。第二,有些药品或医疗器械在海外已上市,但在日本尚未获批。第三,在一般医疗现场,非已上市获批适应症的药品・医疗器械。IIT的目的:协助企业把未能积极开发的药品批准上市;缩小与海外的差距;提高国民的医疗水平和增加选择;进入医保减轻患者经济负担。
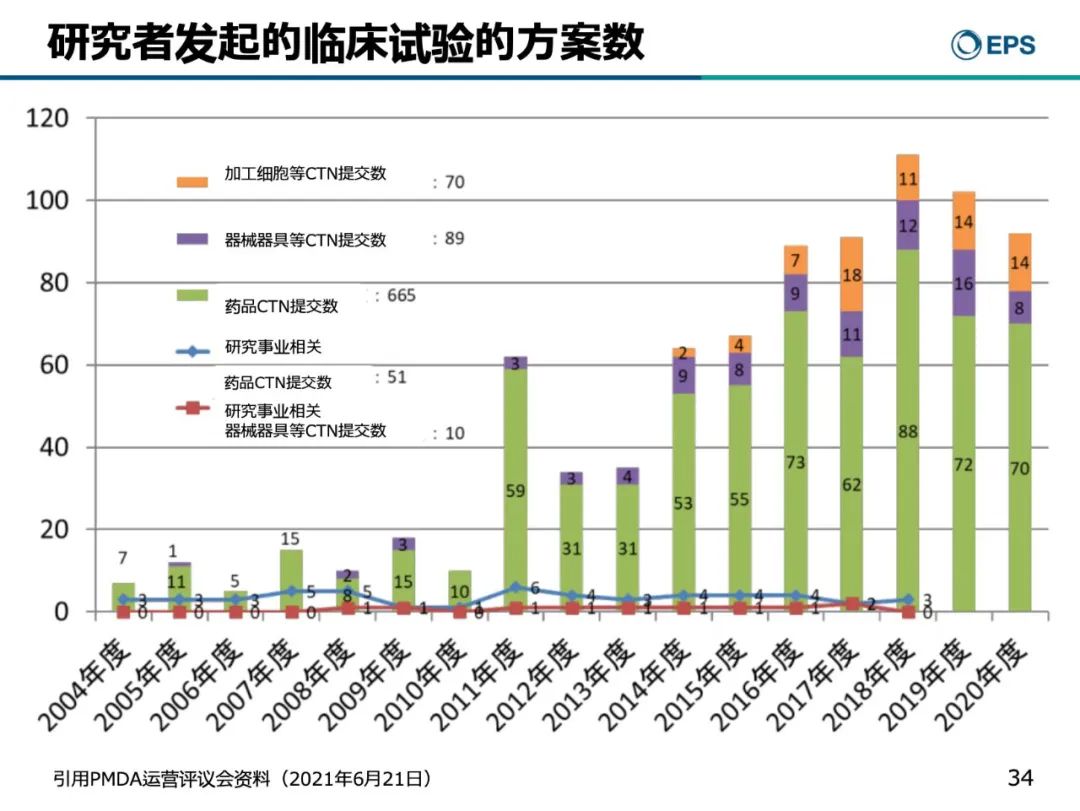
据PMDA统计,2004年-2020年,日本IIT的药品临床试验数量逐年增加, 从2016年开始增加速度非常地迅猛,2018年甚至达到了最高峰的114件。(如下图)。
随着IIT项目的快速增长,另外一个很重要的议题是,那么IIT的资金来源是什么?在日本,IIT研究经费通常来自公众资金,AMED国立开发研究法人和日本医疗研究开发机构(https://www.amed.go.jp/)以及来自制药公司的民间赞助。
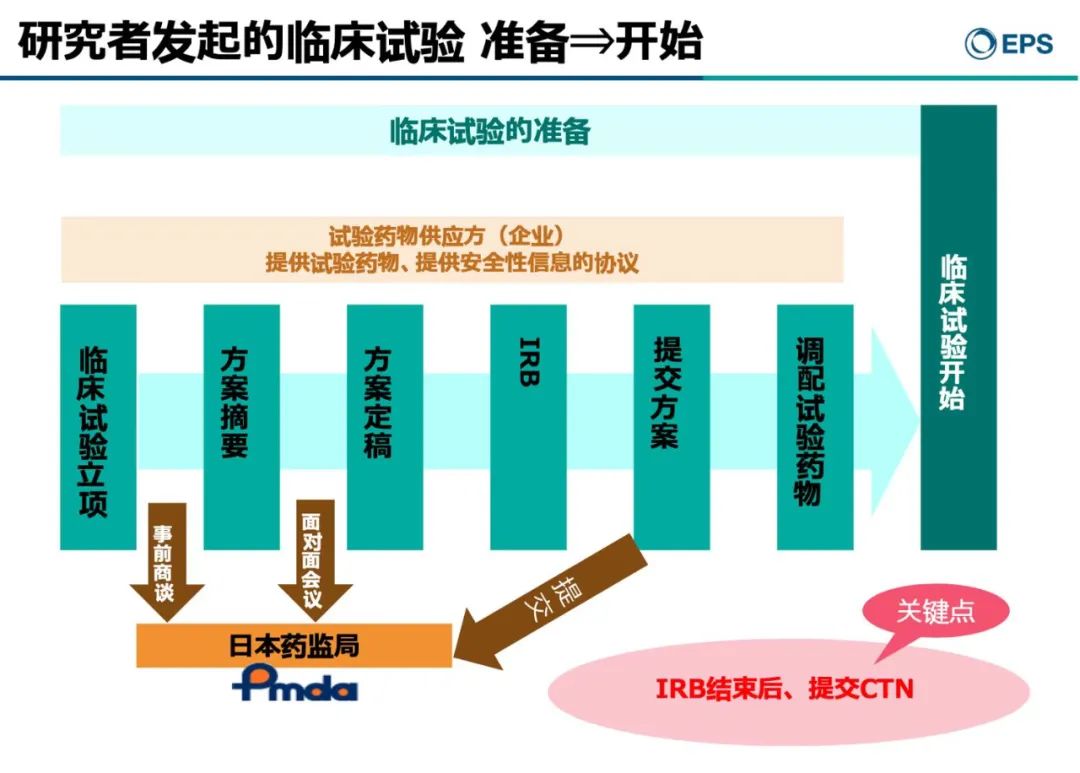
IIT的流程与注册临床试验大致一样,需要临床试验立项、准备阶段。在试验开展前需要做好整体计划,包括撰写临床试验相关资料,通过IRB审查,提交CTN,向临床研究中心发放试验药物等。森冈久代女士强调说,即便是IIT,日本的研究者也恪守ICH GCP以及日本国内的J-GCP,最大的区别为IIT是IRB结束后,提交CTN。
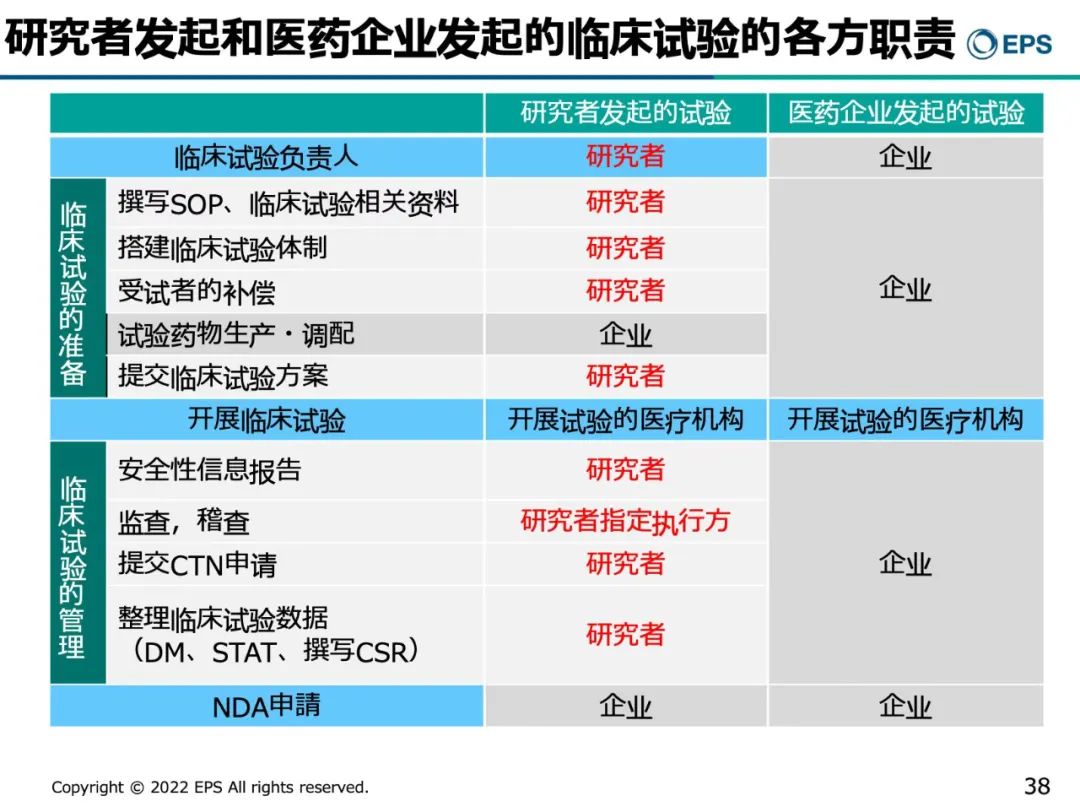
在日本,IIT和注册临床试验都有明确的职责,制药企业发起的注册临床试验,制药企业需要承担除了执行临床试验以外的所有事务;如果是IIT, 研究者除了需要履行PI的职责外,还要确认SOP, 撰写与临床试验相关的资料,建立项目体制,受试者补偿和保险,提交CTN等都是由研究者来承担。
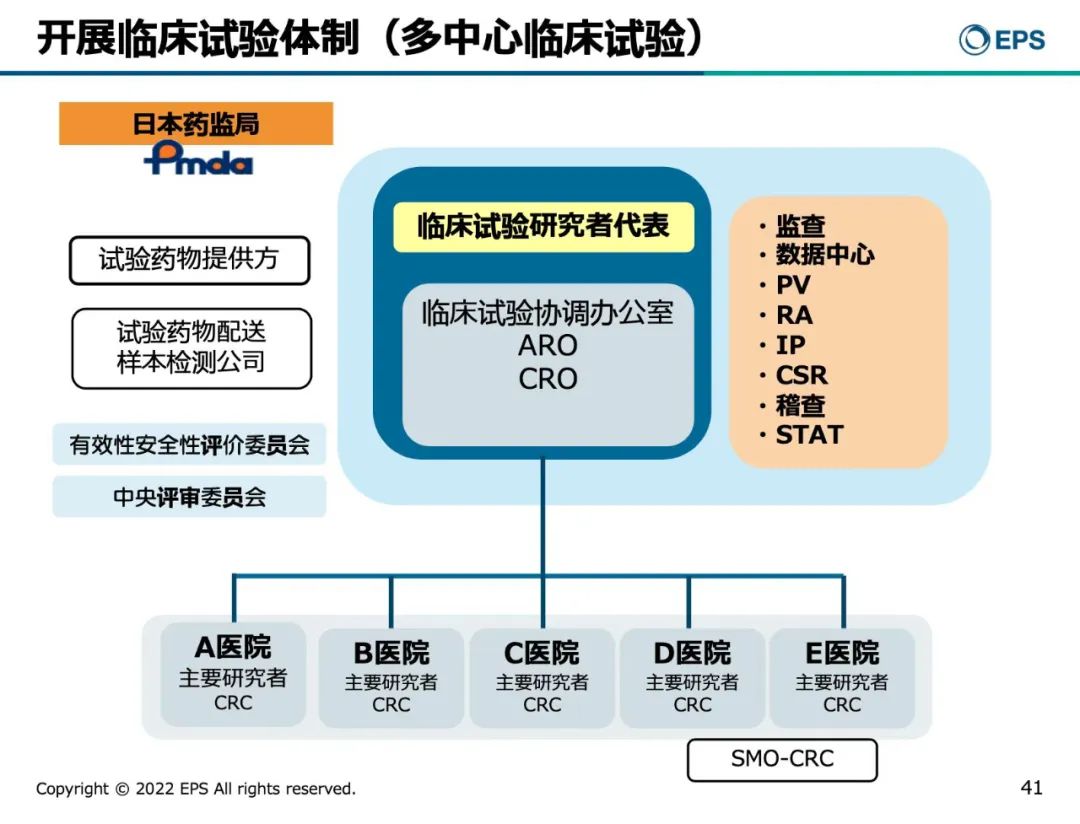 最后,森冈久代女士总结道,研究者发起和制药企业发起的临床试验的差异包括以下3点:
最后,森冈久代女士总结道,研究者发起和制药企业发起的临床试验的差异包括以下3点:
1. 提交临床试验方案步骤 。IIT是在IRB批准后提交CTN,而制药企业发起的临床试验则是提交CTN后30天默认期没有收到PMDA的修改意见后,提交IRB申请。
2. 临床试验监查。在IIT临床试验的监查工作,通常由PI指定CRAs,并以第三方执行,类似稽查的职责,向IRB提交监查报告和接受审核。而制药企业发起的临床试验的监查工作,通常由制药公司安排CRAs。
3. 减轻受试者经济负担。
IIT临床试验可以不支付受试者补偿, 而制药企业发起的临床试验通常需要给7,000~10,000日元的受试者补偿(约合365~520元人民币)。
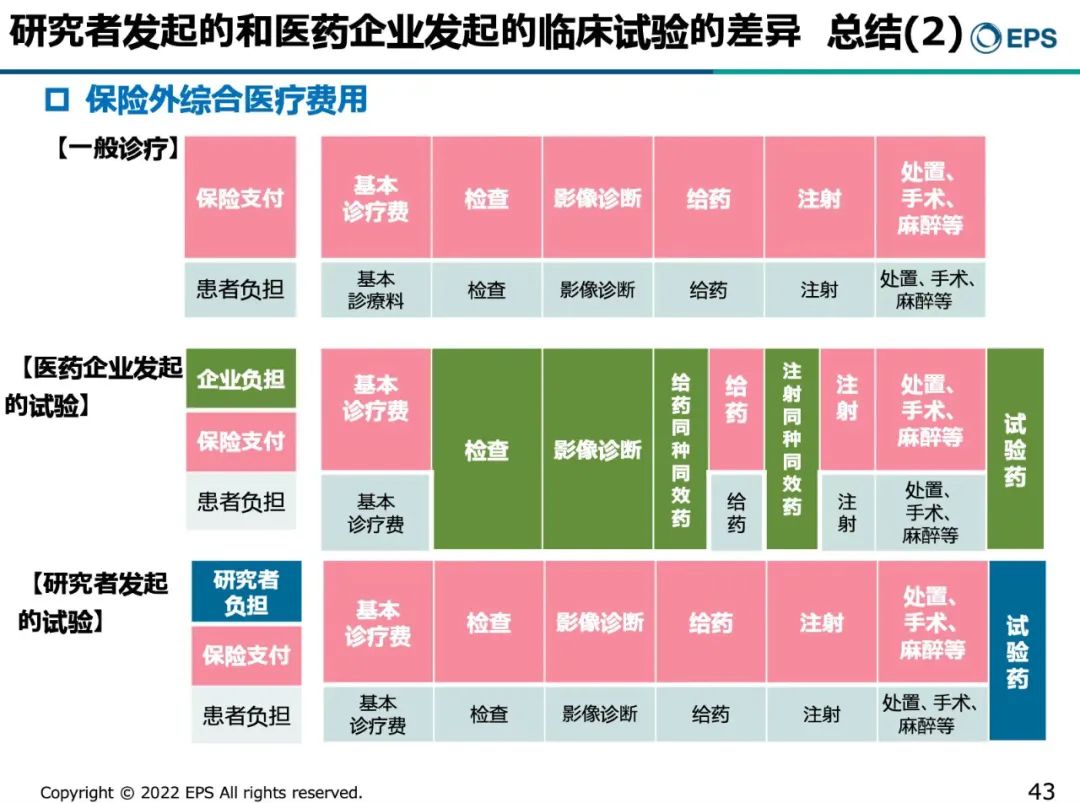
日本是全民医疗保险,但对于临床试验中发生的保险以外的综合医疗费用,无论是IIT还是制药公司发起的临床试验都有清晰规定。通常,IIT的大部分费用由医疗保险支付,只有试验药部分由研究者负担。
结论
综上所述,日本的临床研究和IIT十分发达。笔者通过了解分析原因如下:首先,日本的研究者大多拥有多年扎实的医学教育背景,积累了丰富的临床实践经验,熟悉GCP法规;第二,研究者不但对日常诊疗工作认真细致一丝不苟,还对临床研究充满热情,希望从研究中发现新的治疗手段;第三,研究型医院的现场管理得当,配备人力资料相对充足,有利于项目保质保量的开展和完成。
2021年1月,东京大学医科学研究所附属医院脑肿瘤外科医生藤堂具纪教授主导发起了第一三共制药的溶瘤病毒药物teserpaturev(G47∆,DS-1647)用于治疗恶性、复发性神经胶质母细胞瘤患者的Ⅱ期单臂临床试验。该试验从临床前到实施,完全由藤堂具纪教授团队主导,是日本药品开发史上由研究者发起的创新药开发的成功案例。
这一案例说明,IIT对于新药开发和已上市药品新适应症的拓展具有重要的辅助作用,原因在于:研究者能敏锐发现来自临床的需求开展试验;IIT的试验设计更公正、客观和不偏移,不会倾向于某一申办方,更具有研究者独立的治学思想。如果试验过程依照ICH GCP准则,研究数据真实可靠,研究者发起试验后会充分与试验药研制的药厂合作,最后将IIT研究结果用于注册申报。
近年来,我国IIT的发展日益蓬勃,尤其是新冠疫情以来,各大临床研究机构的研究者纷纷发起相关临床试验。正如广东省人民医院广东省肺癌研究所名誉所长吴一龙教授曾说,IIT需要得到重视,这是发挥研究者主观能动性和创造力的领域。不过,我国对IIT的监管仍处于初步发展阶段,2021年9月9日,国家卫健委正式发布了《医疗卫生机构开展研究者发起的临床研究管理办法(试行)》(以下简称《临床研究管理办法》,并在该年10月1日在北京市、上海市、广东省和海南省先行试点。
《临床研究管理办法》对我国IIT的定义、责任主体有清晰的规定,特别强调所有临床研究均应通过科学审查和伦理审查。医疗卫生机构及研究者要充分尊重受试者的知情权与选择权。医疗卫生机构及其研究者开展临床研究应取得法律法规要求的临床资质,具备相应的能力和必要的资金保障。同时,该《临床研究管理办法》还对研究分类、组织管理、立项管理、财务管理、实施管理、监督管理等均有具体详尽的规定,是我国IIT管理一次重大的里程碑。中山大学肿瘤医院临床研究方法学教研室主任洪明晃教授说,这将令临床研究有法可依,敦促研究者对临床研究的科学性、伦理合规性负责,有助于推进药物研究的深度和广度,为循证医学及新药研发提供可靠依据。不过,仍需要更多医院以此规定具体实践,为此需要来自研究者、监管部门和患者的通力合作。
关于日本的《药品医疗器械法》
日本众议员最早在2002年7月通过《药事法》(Pharmaceutical Affairs Law),该法对日本药品的研发、生产、销售环节实施管理,其目的在于保证药品、医疗器械和化妆品的质量,保护和促进公众健康。2013年,日本将这一《药事法》修改为《药品医疗器械法》(Pharmaceutical and Medical Devices Law,PMDL),将医疗器械的监管纳入新法,并在2013年通过,于2014年11月25日正式实施。
关于日本先驱审查制度
该制度由PMDA在2015年4月建立,旨在将全球开发的创新药、医疗器械和再生医疗产品更早带到日本。特别是在早期临床试验已体现出显著疗效的药物,先驱审查将促进其更早商业化。先驱审查制度的药品需满足如下要求:1.治疗和诊断方法的突破性与革新性;2.针对重大严重影响生命质量的疾病或无法根治的疾病;3.对无有效治疗/诊断方法,或预计其效果较已有治疗/诊断方法有改善,包括安全性的显著提高;4.具有先于全球、在日本尽快进行研发和申请意愿(包括同时申报)的品种。先驱审查制度的时限为6个月,比过往减少一半。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#日本#
90
#真实世界#
105