最强PCR原理攻略
2023-02-12 检验之声 检验之声 发表于安徽省

PCR全称多聚酶链式反应:是一种非常强大的技术,可以通过一种非常简单但是高效的方法来复制DNA,它可看作是生物体外的特殊DNA复制,PCR的最大特点是能将微量的DNA大幅增加。
一.PCR简介
生物学可以被划为两个时代:一个没有PCR,一个有PCR。没有PCR(聚合酶链式反应,Polymerase Chain Reaction)技术,就没有现代分子生物学。PCR技术发明者Kary Mullis,在1993年获得化学诺贝尔奖,世界称之为PCR教父。
生物体的基因组存储在DNA分子内部,但是分析这种遗传信息需要大量的DNA。1985年,Kary Mullis发明了一种有效的方法,该方法可在短时间内大量复制少量DNA。通过加热,DNA分子的两条链被分离,并且已添加的DNA构件与每一条链结合。借助酶DNA聚合酶,可以形成新的DNA链,然后可以重复该过程。PCR在医学研究和法医学领域都具有重要意义。
二.PCR是干什么的?
PCR全称多聚酶链式反应(polymerase chain reaction),是一种非常强大的技术,可以通过一种非常简单但是高效的方法来复制DNA,它可看作是生物体外的特殊DNA复制,PCR的最大特点是能将微量的DNA大幅增加。
三.PCR的原理是什么?
我们知道DNA复制时,以亲代DNA的两条链分别作为模板,在DNA聚合酶的催化下,按碱基互补的原则合成两条与模板链互补的新链,组成新的DNA分子,新形成的两个子代DNA与亲代DNA的碱基顺序完全一样。由于子代DNA分子中一条链来自亲代,而另一条链是新合成的,因此这种复制方式称为半保留复制。
用简单的方式来理解PCR就像是 “心灵手巧的小姑娘学习织围巾” 在织围巾(PCR)的过程中,首先拆解原始织样得到元素织样(模板DNA)、起针(引物对)、毛线(dNTP)和针(Taq酶以及缓冲液体系)串起来,让DNA分子无处可逃。
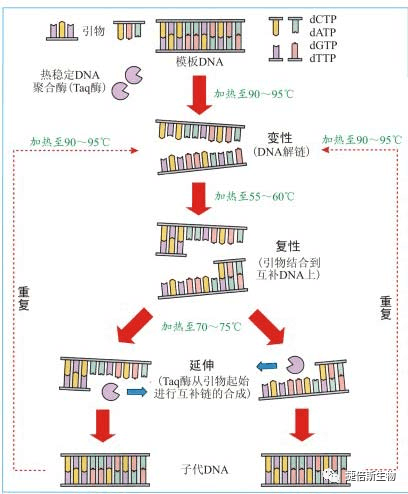
四.PCR实验基本步骤
PCR反应准备工作
1.PCR反应体系
-
10×扩增缓冲液 10μl
-
4种dNTP混合物(终浓度) 各100~250μmol/L
-
引物(终浓度) 各5~20μmol/L
-
模板DNA 0.1~2μg
-
Taq DNA聚合酶 5~10 U
-
Mg2+(终浓度) 1~3mmol/L
-
补加双蒸水 100 μl
其中dNTP、引物、模板DNA、Taq DNA聚合酶以及Mg2+的加量(或浓度)可根据实验调整,以上数据仅供大致参考值。
PCR反应五要素:
-
引物:(PCR引物为DNA片段,细胞内DNA复制的引物为一段RNA链)引物有多种设计方法,由PCR在实验中的目的决定,但基本原则相同。
-
酶:PCR所用的酶主要有两种来源:Taq和Pfu,分别来自两种不同的噬热菌。其中Taq扩增效率高但易发生错配。Pfu扩增效率弱但有纠错功能。所以实际使用时根据需要必须做不同的选择。
-
dNTP包括dATP, dGTP, dTTP, dCTP
-
模板:模板即扩增用的DNA,可以是任何来源,但有两个原则,第一纯度必须较高,第二浓度不能太高以免抑制。
-
缓冲液:(其中需要Mg2+)
缓冲液的成分最为复杂,除水外一般包括四个有效成分:
缓冲体系,一般使用HEPES或MOPS缓冲体系;
一价阳离子,一般采用钾离子,但在特殊情况下也可使用铵根离子;
二价阳离子,即镁离子,根据反应体系确定,除特殊情况外不需调整;
辅助成分,常见的有DMSO、甘油等,主要用来保持酶的活性和帮助DNA解除缠绕结构。
2.PCR引物设计
PCR反应中有两条引物,即5′端引物和3′引物。设计引物时以一条DNA单链为基准(常以信息链为基准),5′端引物与位于待扩增片段5′端上的一小段DNA序列相同;3′端引物与位于待扩增片段3′端的一小段DNA序列互补。
-
引物设计的基本原则
-
引物长度:15-30bp,常用为20bp左右。
-
引物碱基:G+C含量以40-60%为宜,G+C太少扩增效果不佳,G+C 过多易出现非特异条带。ATGC最好随机分布,避免5个以上的嘌呤或嘧啶核苷酸的成串排列参照。
-
引物内部不应出现互补序列。
-
两个引物之间不应存在互补序列,尤其是避免3 ′端的互补重叠。
-
引物与非特异扩增区的序列的同源性不要超过70%,引物3′末端连续8个碱基在待扩增区以外不能有完全互补序列,否则易导致非特异性扩增。
-
引物3‘端的碱基,特别是最末及倒数第二个碱基,应严格要求配对,最佳选择是G和C。
-
引物的5′端可以修饰。如附加限制酶位点,引入突变位点,用生物素、荧光物质、地高辛标记,加入其它短序列,包括起始密码子、终止密码子等。
3.模板的制备
PCR的模板可以是DNA,也可以是RNA。模板的取材主要依据PCR的扩增对象,可以是病原体标本如病毒、细菌、真菌等。也可以是病理生理标本如细胞、血液、羊水细胞等。法医学标本有血斑、、毛发等。
标本处理的基本要求是除去杂质,并部分纯化标本中的核酸。多数样品需要经过SDS和蛋白酶K处理。难以破碎的细菌,可用溶菌酶加EDTA处理。所得到的粗制DNA,经酚、氯仿抽提纯化,再用乙醇沉淀后用作PCR反应模板。
4.反应的控制
-
PCR反应的缓冲液 提供合适的酸碱度与某些离子
-
镁离子浓度 总量应比dNTPs的浓度高,常用1.5mmol/L
-
底物浓度 dNTP以等摩尔浓度配制,20~200umol/L
-
TaqDNA聚合酶 2.5U(100ul)
-
引物 浓度一般为0.1 ~ 0.5umol/L
-
反应温度和循环次数
-
变性温度和时间 95℃,30s
-
退火温度和时间 低于引物Tm值5 ℃左右,一般在45~55℃
-
延伸温度和时间 72℃,1min/kb(10kb内)
-
Tm值=4(G+C) +2(A+T)
循环次数 :一般为25 ~ 30次。循环数决定PCR扩增的产量。模板初始浓度低,可增加循环数以便达到有效的 扩增量。但循环数并不是可以无限增加的。一般循环数为30个左右,循环数超过30个以后,DNA聚合酶活性逐渐达到饱和,产物的量不再随循环数的增加而增加,出现了所谓的“平台期”
循环参数设置
1.预变性:模板DNA完全变性与PCR酶的完全激活对PCR能否成功至关重要,建议加热时间参考试剂说明书,一般未修饰的Taq酶激活时间为两分钟。
2.变性步骤:循环中一般95℃,30秒足以使各种靶DNA序列完全变性,可能的情况下可缩短该步骤时间。变性时间过长损害酶活性,过短靶序列变性不彻底,易造成扩增失败。
3.引物退火:退火温度需要从多方面去决定,一般根据引物的Tm值为参考,根据扩增的长度适当下调作为退火温度。然后在此次实验基础上做出预估。退火温度对PCR的特异性有较大影响。
4.引物延伸:引物延伸一般在72℃进行(Taq酶最适温度)。但在扩增长度较短且退火温度较高时,本步骤可省略延伸时间随扩增片段长短而定,一般推荐在1000bp以上,含Pfu及其衍生物的衍生设定为1min/kbp。
5.循环数:大多数PCR含25-35循环,过多易产生非特异扩增。
6.最后延伸:在最后一个循环后,反应在72℃维持10-30分钟.使引物延伸完全,并使单链产物退火成双链。
PCR反应步骤
标准的PCR过程分为三步:
DNA变性:(90℃-96℃):双链DNA模板在热作用下,氢键断裂,形成单链DNA
退火:(60℃-65℃):系统温度降低,引物与DNA模板结合,形成局部双链。
延伸:(70℃-75℃):在Taq酶(在72℃左右,活性最佳)的作用下,以dNTP为原料,
从引物的3′端开始以从5′→3′端的方向延伸,合成与模板互补的DNA链。每一循环经过变性、退火和延伸,DNA含量即增加一倍。
如图所示:现在有些PCR因为扩增区很短,即使Taq酶活性不是最佳也能在很短的时间内复制完成,因此可以改为两步法,即退火和延伸同时在60℃-65℃间进行,以减少一次升降温过程,提高了反应速度。
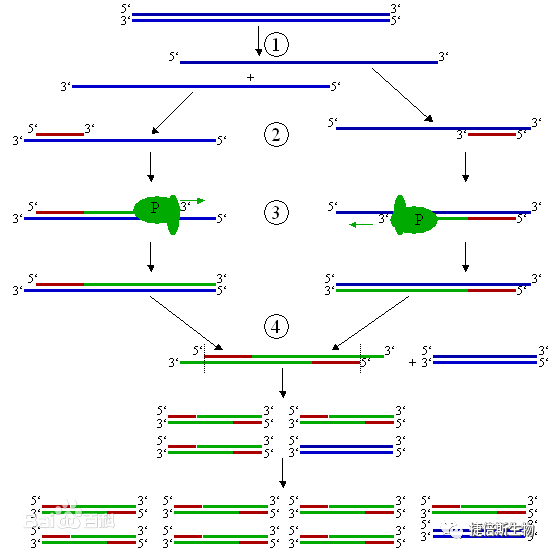 PCR反应特点
PCR反应特点
1.特异性强
PCR反应的特异性决定因素为:
①引物与模板DNA特异正确的结合;
②碱基配对原则;
③Taq DNA聚合酶合成反应的忠实性;
④靶基因的特异性与保守性。
2.灵敏度高
3.简便、快速
4.纯度要求低
PCR反应结果的检测
PCR反应扩增出了高的拷贝数,下一步检测就成了关键。荧光素(溴化乙锭,EB)染色凝胶电泳是最常用的检测手段。电泳法检测特异性是不太高的,因此引物两聚体等非特异性的杂交体很容易引起误判。但因为其简捷易行,成为了主流检测方法。近年来以荧光探针为代表的检测方法,有逐渐取代电泳法的趋势。
常见问题
1.假阴性
不出现扩增条带。PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及溴乙锭的使用, ④PCR循环条件。寻找原因亦应针对上述环节进行分析研究。
模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。⑤模板核酸变性不彻底。在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应 固定不宜随意更改。
2.阴性
需注意的是有时忘加Taq酶或溴乙锭。引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不理想、容易弥散的常见原因。
3.假阳性
出现的PCR扩增条带与目的靶序列条带一致,有时其条带更整齐,亮度更高。
引物设计不合适:选择的扩增序列与非目的扩增序列有同源性,因而在进行PCR扩增时,扩增出的PCR产物为非目的性的序列。靶序列太短或引物太短,容易出现假阳性。需重新设计引物。
靶序列或扩增产物的交叉污染:这种污染有两种原因:一是整个基因组或大片段的交叉污染,导致假阳性。这种假阳性可用以下方法解决:操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外。除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管及样进枪头等均应一次性使用。必要时,在加标本前,反应管和试剂用紫外线照射,以破坏存在的核酸。二是空气中的小片段核酸污染,这些小片段比靶序列短,但有一定的同源性。可互相拼接,与引物互补后,可扩增出PCR产物,而导致假阳性的产生,可用巢式PCR方法来减轻或消除。
出现非特异性扩增带:PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带 与非特异性扩增带。非特异性条带的出现,其原因:一是引物与靶序列不完全互补、或引物聚合形成二聚体。二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。其对策有:必要时重新设计引 物。减低酶量或调换另一来源的酶。降低引物量,适当增加模板量,减少循环次数。适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。
出现片状拖带或涂抹带:PCR扩增有时出现涂抹带或片状带或地毯样带。其原因往往由于酶量过多或酶的质量差,dNTP浓度过高,Mg2+浓度过高,退火温度过低,循环次数过多引起。其对策有:减少酶量,或调换另一来源的酶。②减少dNTP的浓度。适当降低Mg2+浓 度。增加模板量,减少循环次数。
实验所需试剂
| 货号 | 试剂名称-(GBCBIO) |
| R1105 | TRNsol/TRIzol RNA提取试剂 |
| D2105 | 组织/细胞DNA提取试剂盒 |
| D5105 | Hpure 植物DNA提取试剂盒 |
| D3105 | 细菌DNA提取试剂盒 |
| P3711 |
Long Taq DNA聚合酶 |
| P3511 | Taq DNA聚合酶 |
| D2314 | dNTP Mix |
| G3410 |
电泳缓冲液10 x Tris-MOPS-SDS Running Buffer |
| T2866 | Tris-硼酸-EDTA 缓冲液|5×TBE Buffer |
| G3470 | 电泳缓冲液10 x Tris-MES-SDS Running Buffer |
| G4566 | Tris-乙酸-EDTA缓冲液|50XTAE |
| 0288 | 氯化镁 | Magnesium Chloride, Hexahydrate |
| M3705 | DNA分子标准物DNA Marker IV|DNA Marker IV |
| G4550 |
6XDNA上样缓冲液|6XDNA Loading Buffer |
| P3411 | DNA扩增2xPCRmix Master|2xPCRmix Master |
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



