
患者男性,27岁。以“体检发现胰腺占位性病变2月”入院。
体检:面部色素沉积,皮肤黯黑、皮肤巩膜无黄染。既往无糖尿病史、高血压病史。无外源性激素服用史。全腹CT平扫及增强见胰头部一混合回声病灶,约119 mm×63 mm;双侧肾上腺形态饱满(图1A)。
实验室检查:
尿游离皮质醇58184.16nmol/24h;
8点时促肾上腺皮质激素(adreno cortico tropic hormone, ACTH)6.04pmol/L,16点时5.32pmol/L,24点时4.26pmol/L;
上午皮质醇>1655.31nmol/L,下午也>1655.31nmol/L;
胰岛素0分时101.70pmol/L,60分时372.80pmol/L,120分是342.50pmol/L;
C-肽0分时1.06nmol/L,60分时2.01nmol/L,120分时2.16nmol/L;
促甲状腺0.437uIU/ml,游离T3 2.09pmol/L,游离T4 8.09pmol/L。
头颅磁共振示鞍内垂体高径约0.5cm,增强扫描垂体信号均匀,垂体柄居中;视交叉的形态、走形未见异常;双侧海绵窦未见异常信号(图1B)。
肺部CT未发现异常。进一步完善肾上腺多排CT扫描示双侧肾上腺增粗,形态欠规则。
临床表现为顽固性低钾,给予持续性静脉钾离子泵入。经多学科会诊考虑胰腺神经内分泌肿瘤致异位ACTH综合征。择期行胰十二指肠切除术,术后病理报告:考虑胰腺腺泡细胞癌,伴出血、坏死及囊性变,形态学考虑神经内分泌肿瘤(G3)可能性大。免疫组化染色:CgA(-),Syn(-),Ki67(+),AACT(+), CK8(+),CK18(+),ACTH(-),CD56(散在+),核分裂象3~6个/10HPF(图2)。胰十二指肠切除术后复查皮质醇指标,皮质醇上午为422.19nmol/L,下午为987.76nmol/L,尿24小时皮质醇10889.10nmol/L,较术前均有下降。术后无出血及胰漏、胆漏等并发症,但出现发热,腹腔感染,肺部感染及Ⅰ型呼吸衰竭。给予积极抗炎及呼吸机辅助呼吸对症治疗,但由于经济原因患者最终选择出院。

图1 腹部增强CT见胰头部占位(1A),头颅磁共振未见明显异常(1B)
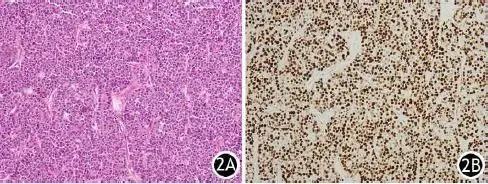
图2 肿瘤组织病理改变(2A, HE ×400)及Ki67阳性表达(2B,免疫组化染色 ×200)
讨论
库欣综合征又称皮质醇增多症,是由于多种因素引起肾上腺皮质长期分泌过量皮质醇而产生的一系列临床症状,表现为满月脸、多血质外貌、向心性肥胖、痤疮、紫纹、高血压、继发性糖尿病和骨质疏松等。异位ACTH综合征是库欣综合征的病因之一,是指由垂体以外的肿瘤过度分泌ACTH而促使其靶腺增生并分泌大量皮质醇而引起的一种综合征。异位ACTH病因不明,多发生于胸腔,见于小细胞肺癌、支气管癌等患者,胰腺来源的少见。查阅万方及知网数据库,国内只报道2例胰腺神经内分泌肿瘤合并异位ACTH综合征患者,临床表现为顽固性低钾、水样便及高血糖等症状。国外文献检索到3例报告,主要为儿童、青少年。本例为中年男性,存在库欣综合征面貌特征,术前检查伴有昼夜皮质醇节律消失,尿皮质醇明显升高,可诊断为库欣综合征,并且ACTH指标未受到抑制,根据影像学检查垂体、肺部、双侧肾上腺均未见异常,仅发现胰头部肿物占位,并伴有异位库欣常见临床表现,即尿皮质醇极度升高,顽固性低钾、代谢性碱中毒等,术后复查血及尿皮质醇指标均较术前下降。因此本例异位ACTH综合征的病因考虑为胰头部肿瘤造成。
对胰腺腺泡细胞癌伴异位ACTH综合征的患者,联合放化疗有一定的疗效,可以使血清ACTH及皮质醇激素逐渐降至正常,但该病预后欠佳。由于该类型病理的形态学表现类似神经内分泌肿瘤,同时往往伴有顽固性低钾、腹泻等临床内分泌综合征的表现而被误诊,因此在临床工作中遇到临床内分泌综合征的患者也需要考虑到该病的可能。文献也显示儿童胰母细胞瘤也可表现为ACTH综合征。
全文刊载于
《中华胰腺病杂志》
2020年 第20卷 第3期 243-244页
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#细胞癌#
74
#综合征#
71
学习了
132