
柳叶刀重磅综述:B细胞非霍奇金淋巴瘤
2024-04-17 聊聊血液 聊聊血液 发表于陕西省

近日《柳叶刀》再次发表B-NHL综述,概述了B-NHL的病理生物学、分类和预后,总结了目前关于成熟 B 细胞非霍奇金淋巴瘤最常见亚型的生物学和临床治疗,还强调了分子学方面的新发现,以及新的治疗手段。
B细胞非霍奇金淋巴瘤
在发达国家,B 细胞非霍奇金淋巴瘤(B-NHL)的发病率为20例新发病例/10万人/年。B-NHL可累及任何器官,具有异质性致病机制、临床表现和病程,从无症状、惰性至侵袭性极强不等。CD20单抗联合常规化疗仍是大多数B-NHL亚型的标准治疗,大多数患者可获得持久的疗效;但高危患者容易早期复发,临床预后很差。
《柳叶刀》杂志上次是在2017年发表NHL的综述,从那之后,对该异质性恶性肿瘤群体的生物学背景、新诊断方法的可用性有了更深入的了解,此外新的靶向和免疫治疗方法的开发和实施提高了治疗能力。从而为B-NHL的诊断、预后和治疗管理方面的新发展铺平了道路,可以实现更好和个性化的疾病管理。过去5年中最重要的突破包括诊断恶性淋巴瘤修订分类,确定预后相关的遗传学变异,以及批准使用免疫系统靶向肿瘤的治疗手段,主要是过继T细胞治疗,从而扩大了治疗范围并改善了治疗效果,特别是对于复发患者。
近日《柳叶刀》再次发表B-NHL综述,概述了B-NHL的病理生物学、分类和预后,总结了目前关于成熟 B 细胞非霍奇金淋巴瘤最常见亚型的生物学和临床治疗,还强调了分子学方面的新发现,以及新的治疗手段,特别是靶向免疫系统的疗法,如CAR-T细胞治疗和双特异性抗体。现翻译全文,水平有限,如有错误敬请谅解。

病理学及分类
非霍奇金淋巴瘤包括淋巴组织的多种恶性肿瘤,可来源于 B 细胞 (85-90%)、T细胞或NK细胞的克隆扩增。B 细胞淋巴瘤可发生于正常 B 细胞发育的任何阶段,但大多数起源于生发中心,包括 Burkitt 淋巴瘤、生发中心 B 细胞样 (GCB) 亚型弥漫性大B细胞淋巴瘤 (DLBCL) 和滤泡性淋巴瘤。在生发中心反应期间,B细胞 DNA 的两种不同修饰可改变 B 细胞受体:体细胞超突变(somatic hypermutation)和类转变重组(class-switch recombination)。这些过程对于抗体多样性的产生和正常免疫反应至关重要,但它们容易出错,并引起导致淋巴瘤发生的 DNA 损伤。大多数套细胞淋巴瘤来源于 B 细胞,其并非来自生发中心,携带未突变的免疫球蛋白重链可变 (IGHV) 基因。但有研究表明,15-40%的套细胞淋巴瘤携带 IGHV 超突变,表明其起源于已经经历了生发中心转运的细胞。边缘区淋巴瘤主要来源于生发中心后B细胞,但结内和脾边缘区淋巴瘤中的一部分病例具有未突变或最小突变的IGHV,表明可能来源于不依赖于生发中心的细胞。
恶性 B-NHL 细胞的生物学特征通常可反映出淋巴瘤来源的对应健康来源细胞。此外,不同淋巴瘤亚型的不同生物学特征是由其复发性的遗传、表观遗传学和其他分子学异常所引起。关于最常见 B-NHL 病理生物学的具体特征将在相应章节中单独讨论。
2022年公布了两个更新的 B-NHL 分类:成熟淋巴肿瘤国际共识分类 (ICC),是血液病理学会和欧洲血液病理学协会与血液学家、肿瘤学家和科学家(临床顾问委员会)的共同成果;以及 WHO 国际癌症研究机构推广的造血和淋巴系统肿瘤WHO-2022 分类。这两个建议都包括部分亚型命名的修改、诊断标准的细化和新亚型的识别。此外,这两个提案都认识到分子学和基因组数据在某些亚型的诊断和临床决策中越来越重要,但在其他亚型中没有得到充分验证。这两种分类有许多共同的方面,但在术语、诊断标准和特定亚型的考虑方面也存在差异(见panel)。

临床表现、分期和预后
B-NHL 患者通常表现为无痛性淋巴结肿大,有时可合并全身症状,如发热、盗汗、体重减轻或疲乏。但由于淋巴瘤表现可发生于淋巴和造血系统以外的任何器官,因此可能存在广泛的临床表现。
为实现准确诊断,应由血液病理学家使用标准化诊断程序对活检样本进行评估,最好是来自受累淋巴结或其他器官肿瘤的切除活检。定义疾病分期和估计预后的进一步诊断检查应包括精确的病史、体格检查以及实验室和影像学检查。
PET-CT是精确定义疾病分期的最准确的仪器,广泛用于治疗前评估。与 CT 扫描相比,PET-CT分期准确性较高,会导致10-30%的患者降期或升期(后者更常见)。此外,PET-CT分期可实现与治疗结束评估直接比较,其中 PET-CT 是标准手段。因此,根据国际恶性淋巴瘤会议影像学工作组共识,应将 PET-CT 视为FDG高摄取淋巴结淋巴瘤常规分期的金标准(所有组织学亚型,除了皮肤边缘区淋巴瘤、慢性淋巴细胞白血病/小淋巴细胞淋巴瘤外的所有组织学、华氏巨球蛋白血症和蕈样肉芽肿,除非怀疑侵袭性转化)。此外,PET-CT在评估骨髓受累方面也很敏感。在 DLBCL 中,PET-CT阳性骨髓信号足以确定晚期疾病,而额外的骨髓活检是多余的。在滤泡性淋巴瘤中, PET联合骨髓活检在识别骨髓受累方面比任一单独检测的准确性均更高;关于骨髓活检是否可为确定治疗选择提供关键信息,目前仍存在争议。滤泡性淋巴瘤的一项大型多中心、多试验队列的结果认为,骨髓活检在评估缓解或预测无进展生存期或总生存期方面不具有额外价值。因此,仅在可能影响治疗(包括确认局限性分期和评估血细胞减少)的情况下,才建议在首诊时进行骨髓活检。对于治疗结束评估,PET-CT和 Deauville 评分是预测无病生存期的一种非常敏感的工具,尤其是在 DLBCL 中,因此应被视为标准治疗。Deauville 评分是一种基于 FDG 摄取目视解读的5分量表评分系统,评分≤3视为完全缓解,表明长期预后良好。
最常用的分期系统是 Ann Arbor 和 Lugano。Ann Arbor 分期系统最初是为霍奇金淋巴瘤建立,随后扩展到B-NHL。该分期系统基于受累部位的位置和数量和有无全身症状,共定义4个分期。考虑到 III 期和 IV 期接受的治疗多为相似,Lugano出于治疗目的将患者重新分类为局限期(I期和 II 期)或进展期(III期或 IV 期)。
国际预后指数 (IPI) 是首个预测侵袭性 B-NHL 患者预后的国际共识系统,包含以下风险因素:年龄>60岁、III或 IV 期疾病、血清乳酸脱氢酶高于正常值上限、东部肿瘤协作组 (ECOG) 体能状态评分≥2分和至少累及2个结外部位。对于多种惰性淋巴瘤亚型,也开发了调整的预后指数:对于接受利妥昔单抗为基础治疗的临床风险分层,美国国家综合癌症网络国际预后指数 (NCCN-IPI)在区分低危和高危(定义为5年总生存率<50%)DLBCL患者方面优于IPI。滤泡性淋巴瘤国际预后指数 (FLIPI) 是一种5因素模型,包括年龄、分期、血清乳酸脱氢酶、血红蛋白水平和受累淋巴结数量,已在CD20 靶向抗体利妥昔单抗时代得到验证。在套细胞淋巴瘤中,套细胞淋巴瘤国际预后指数 (MIPI) 根据4个独立的预后因素:年龄、体能状态、血清乳酸脱氢酶和白细胞计数,定义了3个风险组。表1比较了几个亚型特异性 IPI 评分。
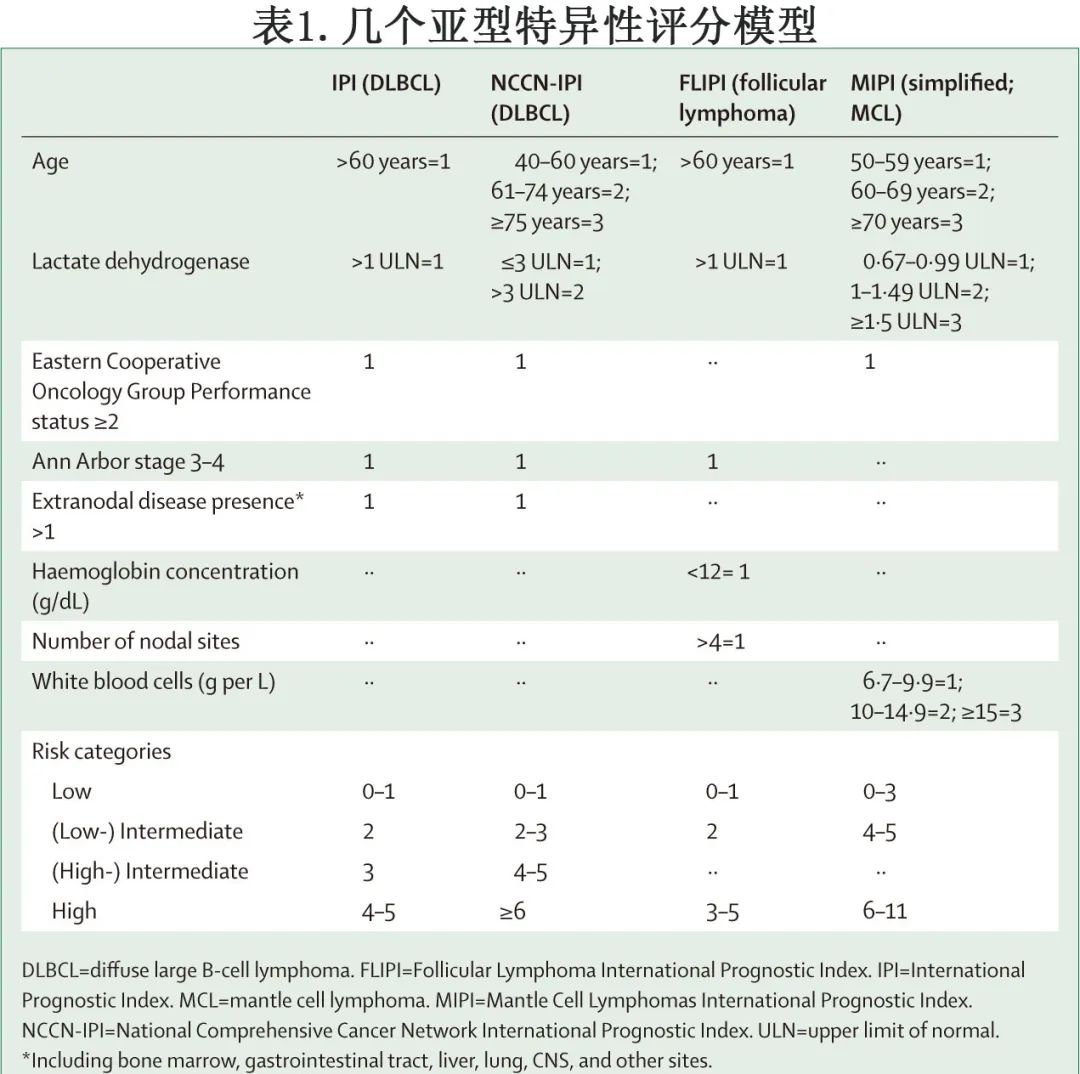
整合临床-遗传因素的更复杂评分目前正在研究中。例如,m7-FLIPI在常用的 FLIPI 评分中增加了7个基因的突变状态(EZH2、ARID1A、MEF2B、EP300、FOX01、CREBBP和CARD11),从而更精确地估计接受免疫化疗患者的5年无失败生存率。此外,在类似患者中还开发了适用于福尔马林固定、石蜡包埋肿瘤活检的基于23个基因表达的无进展生存期稳健预测因子。但这些评分尚未准备好用于常规临床实践。
循环肿瘤 DNA 也是估计预后的一种有前景的候选手段,可用于高危DLBCL、异基因干细胞移植后和 CAR-T 细胞治疗后。
非霍奇金淋巴瘤的特定亚型
华氏巨球蛋白血症(WM)
WM是一种罕见的低度恶性 B 细胞淋巴瘤,定义为小淋巴浆细胞浸润骨髓并存在可检测的血清单克隆IgM。如果不存在骨髓浸润且 IgM 水平低于3g/dL,应诊断为意义未明的 IgM 单克隆丙种球蛋白病。MYD88(L265P) 突变是WM的标志,存在于超过95%的病例中。30-40%的WM患者携带 CXCR4 突变,与无 CXCR4 突变的患者相比,通常与治疗反应和结局较差相关。
大多数患者在诊断时无症状,但部分患者可能发生淋巴结肿大、脾肿大、贫血、神经病变或冷球蛋白血症。约10-15%的患者存在与 IgM 单克隆蛋白高度升高相关的头痛、视物模糊、意识模糊、黏膜出血等高黏滞综合征体征。约1%的患者诊断为 Bing-Neel 综合征,它是WM的一种罕见表现,可累及 CNS。
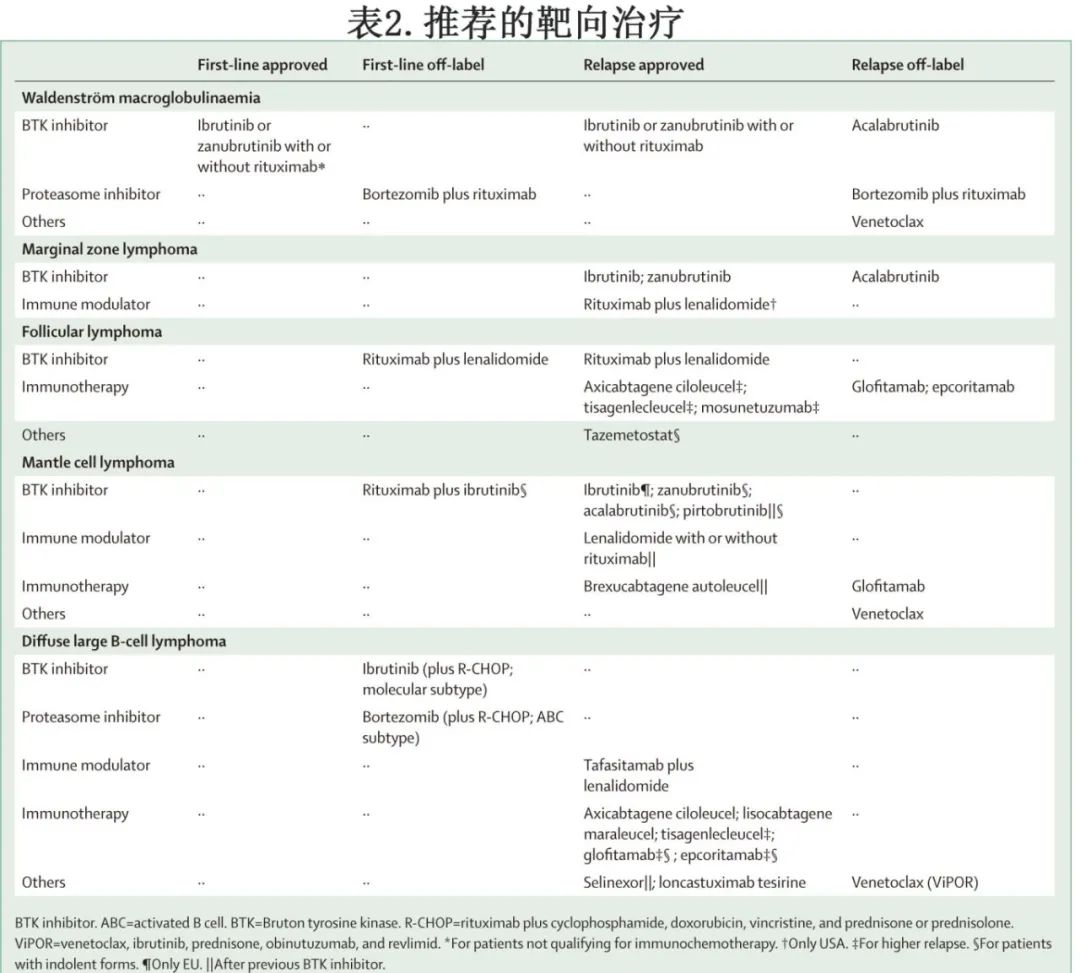
有症状的患者应开始治疗;而无症状患者单纯 IgM 水平升高不应成为开始治疗的指征,除非 IgM 超过60 g/L。在这种情况下,预计患者发生症状性高粘血症的风险相当高,从而可支持在发生不可逆损伤之前治疗无症状患者。烷化剂药物(苯达莫司汀或环磷酰胺)联合利妥昔单抗是一线治疗选择。第一代BTK抑制剂伊布替尼获批用于治疗症状性WM患者,对初治和经治治疗患者均有效,但CXCR4突变患者对伊布替尼的反应率较低。一项随机 III 期研究在150例经治或初治WM中对比伊布替尼联合利妥昔单抗与安慰剂联合利妥昔单抗,结果联合治疗显著改善经治组和初治组的中位无进展生存期,并导致其获批;此外该获益不受MYD88 和 CXCR4 突变状态影响。尽管有效,但伊布替尼治疗与一些毒性作用相关。还评价了二代 BTK 抑制剂阿可替尼和泽布替尼治疗WM;阿可替尼尚未获批,但泽布替尼于2021年在美国和欧洲获批用于既往接受过至少一种治疗的成人WM患者,或作为不适合免疫化疗患者的一线治疗。该批准是基于 ASPEN 研究,这是一项比较伊布替尼与泽布替尼的随机3期研究,报告了两种 BTK 抑制剂的高有效性;但泽布替尼的毒性特征优于伊布替尼,因此应优先使用。对于 BTK 抑制剂治疗后复发的患者,BCL2抑制剂维奈克拉可能是一种有效的挽救治疗选择,但尚未获批。推荐的靶向治疗总结见表2。
边缘区淋巴瘤(MZL)
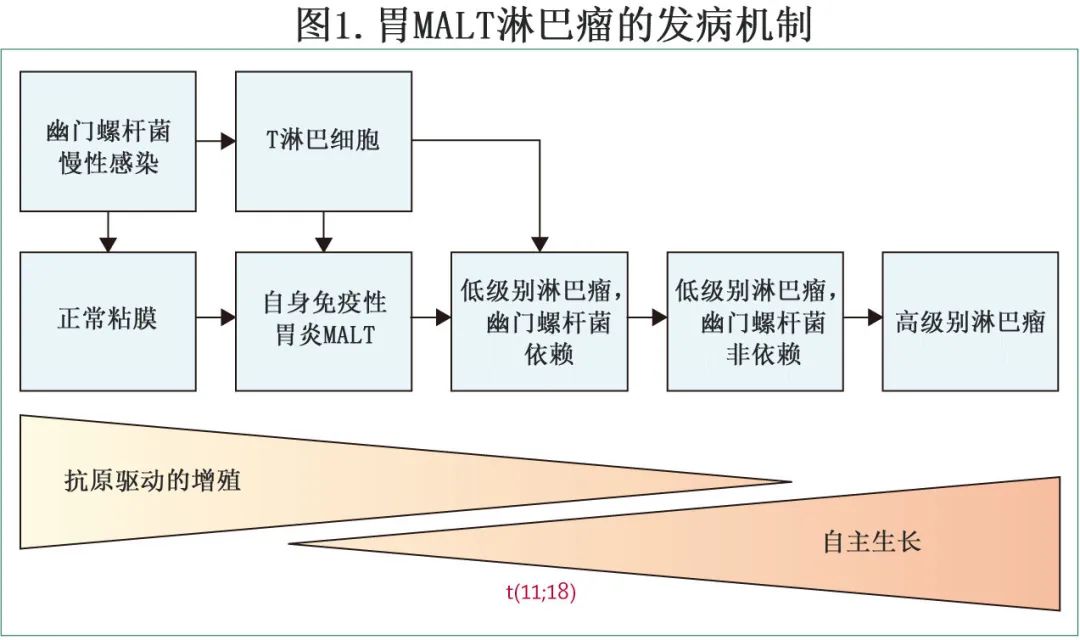
在发达国家,边缘区淋巴瘤约占所有 B-NHL 的5-15%,有三种亚型:结外边缘区淋巴瘤(>60%的病例)脾边缘区淋巴瘤 (20%)和结内边缘区淋巴瘤 (<10%)。结外边缘区淋巴瘤的发病机制似乎是由自身免疫或感染性疾病引起的慢性炎症所启动(图1)。一些基因易位已在结外边缘区淋巴瘤中发现,包括 BIRC3-MALT1 中的t(11;18)(p21;q21)、IGH-MALT1中较罕见的t(14;18)(p32;q21) 和 BCL10-IGH 中的t(1;14)(p22;q32),但在结内边缘区淋巴瘤或脾边缘区淋巴瘤中未发现。结内边缘区淋巴瘤和脾边缘区淋巴瘤的突变谱在KMT2D、NOTCH2、PTPRD、TNFAPI3和 KLF2 中具有共同的改变。
结外边缘区淋巴瘤可发生于任何结外部位,以胃为最常见部位,其次为眼附属器、肺和唾液腺。诊断时可能存在局部淋巴结受累和骨髓浸润,与较差预后相关。为估计预后而开发了结外边缘区淋巴瘤特异性预后指数,可区分3个风险组(年龄≥70岁、Ann Arbor III期或 IV 期和乳酸脱氢酶升高),5年无事件生存率为29-70%。
所有确诊为胃边缘区淋巴瘤并证实幽门螺杆菌感染的患者最初均应接受幽门螺杆菌根除治疗,无论分期如何,多数有临床缓解。然而,t(11;18)(p21;q21) 易位胃边缘区淋巴瘤在胃边缘区淋巴瘤患者中的发生率大约为25%,已知对抗生素治疗耐药;其10年无进展生存率 (26%) 也显著低于无该易位的患者 (57%;p=0.004)。
在重复内镜检查证实淋巴瘤未消退的患者中,受累野放疗可在局限性阶段实现极佳的疾病控制,即使使用 24Gy 的低剂量。对于有症状的晚期患者,利妥昔单抗单药或联合苯丁酸氮芥或苯达莫司汀的全身治疗显示出较好疗效,建议使用。无症状患者可首选观察等待。
脾边缘区淋巴瘤通常累及脾脏、肺门淋巴结、骨髓和血液。约20%的脾边缘区淋巴瘤患者存在自身免疫表现,如自身免疫性溶血性贫血、免疫性血小板减少症、冷凝集素病或获得性血管性血友病等。如果脾肿大出现症状或迅速进展,或者如果观察到任何进行性血细胞减少(血红蛋白<10 g/dL、血小板<80 g/L或中性粒细胞<1g/L),则应开始治疗脾边缘区淋巴瘤。脾切除术不再是首选治疗,已被利妥昔单抗单药治疗广泛取代,后者缓解时间较长。
许多结内边缘区淋巴瘤患者表现为播散性淋巴结肿大和晚期疾病,10-20%的患者存在B 症状。这些病例的治疗应遵循滤泡性淋巴瘤的治疗原则(以下章节中概述)。
对于复发或难治性粘膜相关淋巴组织 (MALT) 淋巴瘤,最新的欧洲肿瘤内科学会指南推荐利妥昔单抗联合免疫调节剂来那度胺作为潜在治疗选择。该建议是基于3期 AUGMENT 研究(利妥昔单抗联合来那度胺 vs 利妥昔单抗联合安慰剂)的结果,导致FDA批准利妥昔单抗联合来那度胺。然而,在63例 MALT 淋巴瘤、脾边缘区淋巴瘤或结内边缘区淋巴瘤患者的子队列中,两种治疗之间未观察到中位无进展生存期的差异。复发性边缘区淋巴瘤的推荐靶向治疗替代方案为 BTK 抑制剂;伊布替尼在63例边缘区淋巴瘤患者中的总缓解率为58%,无进展生存期为15.7个月,并于2017年获批用于治疗需要全身治疗且既往接受过至少一种抗 CD20 治疗的患者。II 期 MAGNOLIA 研究纳入既往接受过至少一种 CD20 靶向治疗的复发性边缘区淋巴瘤患者,对泽布替尼进行评价;泽布替尼的疗效高于伊布替尼,总缓解率为68.2%,估计15个月无进展生存期为82.5%,从而获得FDA批准。在一项2期研究中,阿可替尼治疗复发性边缘区淋巴瘤患者的总缓解率为53%,无进展生存期为27.4个月(12个月无进展生存率为67%);但阿可替尼仍在等待批准。推荐的靶向治疗总结见表2。
滤泡性淋巴瘤(FL)
FL约占所有 B-NHL 病例的25%,是西欧和美国最常见的惰性淋巴瘤。在更新后的分类中,FL亚组的组织学定义发生了一些重大变更:尽管根据 WHO 2016 版中定义的中心母细胞数量,ICC 2022分类仍保留FL的分级(1、2、3A和3B),但新的 WHO-2022 分类用生物学分组取代了该经典分级。大多数滤泡性淋巴瘤 (85%)为WHO分类中所谓的经典型滤泡性淋巴瘤,其特征为多数为滤泡性生长模式、中心细胞和中心母细胞组成以及存在t(14;18)(p32;q21) 易位;此外还定义了两种相关亚型,滤泡性大 B 细胞淋巴瘤和具有少见特征的滤泡性淋巴瘤。滤泡性大 B 细胞淋巴瘤在很大程度上符合滤泡性淋巴瘤 3B 级;该亚型在生物学上有所不同,一般无t(14;18),但表达 CD10 及 p53 和 IRF4/MUM1 表达增加。滤泡性大 B 细胞淋巴瘤预期在临床上表现更像DLBCL,应给予相应治疗。具有少见特征的滤泡性淋巴瘤包括两种不同的亚型:一种具有所谓的母细胞样或大中心细胞细胞学特征,一种以弥漫性生长方式为主。后者命名为 BCL2-R 阴性 CD23 阳性滤泡中心淋巴瘤,在 ICC 中认为是一种特异性滤泡淋巴瘤亚型,缺乏 BCL2-R 并携带 STAT6 突变,导致 CD23 过度表达;该亚型患者经常表现为腹股沟或盆腔大包块,可能具有弥漫性或结节性组织学模式。
除了经典的t(14;18)(p32;q21) 易位外,表观遗传学改变和染色质生物学破坏已证明可驱动滤泡性淋巴瘤发病机制。在绝大多数滤泡性淋巴瘤中发现 KMT2D 或其他组蛋白修饰因子(CREBBP、EZH2、MEF2B和EP300)的突变。
一小部分FL患者诊断为局部低肿瘤负荷(I-II期),24-30 Gy的受累部位放疗(可选择联合利妥昔单抗)是一种潜在的治愈性治疗策略。对于放疗不可行的患者或肿瘤负荷较低的晚期患者,利妥昔单抗单药是推荐选择。在这些病例中,利妥昔单抗的2年维持治疗策略并不优于进展时利妥昔单抗单药再治疗。大多数FL患者表现为晚期 (III 期和 IV 期)。由于目前尚无治愈性治疗策略,临床过程通常为惰性,10-20%自发消退,因此仅应根据是否存在 B 症状、造血功能受损、大包块疾病、重要器官压迫、腹水、胸腔积液或淋巴瘤快速进展等因素开始治疗。当需要治疗时,利妥昔单抗或奥妥珠单抗应与CHOP(环磷酰胺、多柔比星、长春新碱和泼尼松)、苯达莫司汀或CVP(环磷酰胺、长春新碱和泼尼松)等多种化疗方案之一联合使用。一项在晚期初治FL患者中比较利妥昔单抗联合来那度胺与利妥昔单抗联合化疗的随机3期研究显示,两组的疗效相似;但该联合方案尚未获批用于FL的一线治疗。应考虑使用利妥昔单抗或奥妥珠单抗(每2个月一次)维持治疗2年,可改善多种诱导方案后的无进展生存期,但对总生存率无影响。FOLL12 研究比较了标准利妥昔单抗维持治疗与反应适应性(response-adapted)诱导后维持治疗(基于代谢反应和微小残留病分子学评估,使用三种诱导后治疗)。结果显示,与标准利妥昔单抗维持治疗相比,应用代谢学和分子学反应适应性治疗时,3年无进展生存期显著较差。但是应严格评价抗体维持治疗的应用。推荐的靶向治疗总结见表2。
标准免疫化疗诱导后首次缓解持续时间较短(2年内疾病进展 [POD24])是临床结局较差的强预测因素。对于接受非化疗利妥昔单抗双药治疗或利妥昔单抗单药治疗的患者,早期复发的预后也较差。
复发或进展时应进行新的活检,以排除转化为侵袭性 B 细胞淋巴瘤。选择挽救治疗应考虑既往治疗、缓解持续时间和复发时分期。在早期进展 (POD24) 的情况下,可选择非交叉耐药的化疗骨干方案联合利妥昔单抗。对于利妥昔单抗难治性患者或缓解持续时间小于6个月的患者,奥妥珠单抗+苯达莫司汀(+奥妥珠单抗维持治疗)显示可改善既往未暴露于这些药物的患者的中位无进展生存期和总生存期。另外,对于化疗后缓解时间较短的患者,2019年批准的利妥昔单抗联合来那度胺的无化疗方案显示改善缓解率,可视为挽救治疗。可考虑大剂量化疗和自体干细胞移植 (ASCT) 巩固治疗,尤其是对于疑似组织学转化的年轻患者。
新的靶向治疗正在评价,但未在随机3期研究中得到证实。CD19 CAR-T 细胞疗法(axicel 或tisacel)可使难治性FL患者获得持久缓解,从而使 FDA 批准 axicel 和tisacel用于≥二线全身治疗后的复发性FL,包括抗 CD20 单克隆抗体联合烷化剂。在复发或难治性FL患者中开展的一项单臂 II 期研究评估了双特异性抗体mosunetuzumab,这些患者既往接受过≥2线治疗,包括抗 CD20 治疗和烷化剂。结果60%的患者完全缓解,安全性特征可耐受。基于这些结果,mosunetuzumab在欧盟有条件获批用于治疗复发或难治性FL。另外两种双特异性抗体格菲妥单抗(Glofitamab)和 epcoritamab 目前正在临床试验中进行研究,结果令人鼓舞,正在等待批准。EZH2 抑制剂 tazemetostat 是首个获批的靶向表观遗传学的滤泡性疾病治疗药物,对 EZH2 基因激活突变患者(总缓解率69%)和野生型 EZH2 患者(总缓解率35%)均有效。因此,Tazemetostat可用于治疗既往至少接受过两线治疗且携带 EZH2 突变的复发性FL患者,或无其他治疗选择的复发或难治性FL患者。推荐的靶向治疗总结见表2。
套细胞淋巴瘤(MCL)
在发达国家,MCL占恶性淋巴瘤的5-7%。其临床病程具有异质性,介于数年不需要治疗的惰性病程到短期预后不良的高度侵袭性。WHO 2016 年更新的淋巴恶性肿瘤区分了MCL的两种不同亚型:淋巴结MCL(80-90%的病例)的特征为 IGHV 未突变、转录因子性别决定区 Y-box 11(SOX11) 过度表达和通常更具侵袭性的临床病程(图2);白血病型非淋巴结性MCL(10-20%的病例)通常显示IGHV突变、SOX11阴性,并表现为惰性生物学行为(图2)。通过流式细胞术,几乎所有淋巴结MCL患者均有外周血受累;胃肠道息肉病是另一种常见表现,因此淋巴结MCL在其表现上常有结外成分。组织学上,除经典型MCL(有小至中等大小的细胞,核不规则、分裂,染色质致密)外,还有多形性和母细胞样变异。具有母细胞样形态的MCL以类似淋巴母细胞的肿瘤细胞和高增殖为特征,临床病程也更具侵袭性(图2)。
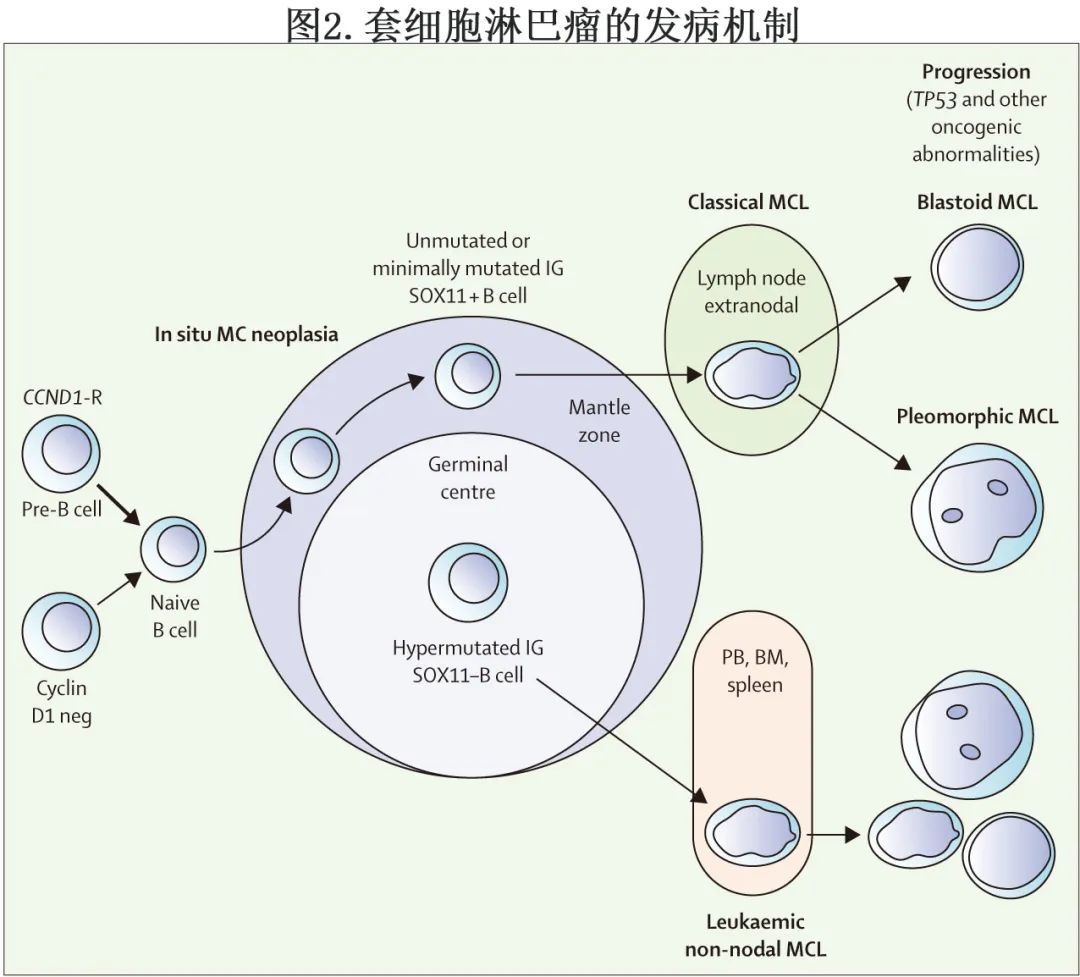
套细胞淋巴瘤的遗传学特征为染色体t(11;14)(p13;q32) 易位,导致细胞周期蛋白 D1 过表达和G1–S期转换时细胞周期失调。免疫组化检测该易位或 cyclin D1 过表达对确诊至关重要。此外检测SOX11 过表达(90%以上的MCL病例存在)也有助于确诊。
二代测序已经开始解开MCL的遗传背景,并确定了众多的复发性体细胞突变,包括参与遗传毒性应激通路的基因(ATM、TP53或CDKN2A)、表观遗传调控因子(NSD2、KMT2D、MEF2B、KMT2C或SMARCA4)以及调节细胞内稳态、细胞生长和细胞死亡的基因(CCND1、TP53、CDKN2A、BIRC3、CARD11、TRAF2、RB1、POT1或NOTCH1/2)。但除了 TP53 外,大多数突变的临床相关性尚不明确。
与较差临床结局相关的重要临床和血清学因素包括:年龄>60岁、体能状态较差、晚期(Ann Arbor III期或 IV 期)、脾肿大、贫血、血清β2-微球蛋白和乳酸脱氢酶水平较高、母细胞样或多形性细胞学、结外表现和全身症状。根据无症状表现、体能状态良好、非淋巴结性病变、血清乳酸脱氢酶正常和低Ki67,部分结局有利的患者子集适合观察等待。目前的证据表明,独立于临床特征的最重要的预后标志物为增殖率、TP53突变或 TP53 高表达。TP53 高表达和 Ki67 >30%,再加上母细胞样形态,可定义为高危生物学,无失败生存期和总生存期显著缩短。在临床环境中,Ki67表达的免疫组化测定是一种可靠的预后标志物,与 MIPI(MIPI-c) 结合时是有用工具,可估计个体风险特征和识别可能适合更侵袭性治疗的高危患者 (Ki67>30%)。
对于≤65岁晚期患者,剂量强化方案是标准治疗,包括免疫化疗诱导序贯大剂量巩固方案和ASCT。但TP53突变的患者并未从这种强化治疗概念中获益,因此应在首次诊断时评价 TP53 状态,以对患者进行相应分层,并应考虑 TP53 突变患者参加一线临床试验。在欧洲,交替应用 R-CHOP 和含阿糖胞苷的 R-DHAP 方案(利妥昔单抗、地塞米松、大剂量阿糖胞苷和顺铂)或序贯 Nordic 方案(利妥昔单抗联合剂量强化 CHOP 和大剂量阿糖胞苷,随后大剂量化疗和ASCT)是常用的诱导策略。但在靶向治疗时代,ASCT的附加价值可能正在下降。在一线和维持治疗的基础上加用伊布替尼,与先进行强化大剂量巩固治疗序贯 ASCT 相比,可改善无失败生存期。对于惰性MCL患者,伊布替尼和利妥昔单抗的一线联合治疗是一种有前景的无化疗方案,可获得较高的完全缓解率和MRD阴性率;但该方案尚未获批。
>65岁不适合移植的患者表现出不同的临床特征,>65岁健康患者应接受常规免疫化疗。在一项大型国际3期研究中,硼替佐米、利妥昔单抗、环磷酰胺、多柔比星和泼尼松 (VR-CAP) 联合治疗优于R-CHOP,尤其是在侵袭性更强的病例中应考虑使用。苯达莫司汀加利妥昔单抗也广泛用于不适合强化治疗方案的患者。3期 SHINE 研究评价了在该联合方案的基础上加用伊布替尼,与苯达莫司汀+利妥昔单抗相比,中位无进展生存期从52.9个月显著改善至80.6个月,但未观察到对总生存期的影响。基于随机研究,利妥昔单抗维持治疗在年轻和老年MCL患者中均显示获益,因此应推荐使用。其他推荐的靶向治疗总结见表2。
对于复发性MCL,已研究多种靶向治疗且结果令人满意。靶向 B 细胞受体通路的 BTK 抑制剂伊布替尼于2013年获批用于复发性MCL,获得显著缓解率,目前正在作为一线治疗的一部分进行评价。此外,另外两种二代 BTK 抑制剂(阿可替尼和泽布替尼)获批用于治疗复发或难治性MCL。维奈克拉单药在高危和伊布替尼治疗后复发患者中获得令人信服的缓解率,正在等待批准。目前正在临床试验中评价维奈克拉联合BTK抑制剂治疗复发或难治性MCL。
免疫治疗方面,自体 CD19 CAR-T brexucabtagene autoleucel实现有前景的结果。中位随访35.6个月,在既往接受过 BTK 抑制剂治疗的复发或难治性MCL患者中,brexucabtagene autoleucel诱导的持久总缓解率为91%,中位无进展生存期为25.8个月,并于2022年获批用于该适应症。正在进行的I期 TRANSCEND B-NHL001 研究 (NCT02631044) 评价第二种 CD19 CAR-T 细胞产品 (lisocel) 治疗复发或难治性MCL。
双特异性抗体目前正在 B 细胞恶性肿瘤中进行研究,在早期研究中接受其治疗的少数MCL患者的结果令人鼓舞。在37例多线治疗MCL患者中进行的1-2期研究中,奥托珠单抗(1000 mg或2000 mg)预处理后评估格菲妥单抗单药治疗,结果诱导较高且持久的完全缓解率(73%)。推荐的靶向治疗总结见表2。
弥漫性大B细胞淋巴瘤(DLBCL)
DLBCL 是最常见和最具侵袭性的 B-NHL 亚型,在发达国家占30-40%。DLBCL 起源于生发中心或活化 B 细胞的克隆性增殖,根据起源细胞 (COO) 亚型可细分为 GCB 样DLBCL、活化 B 细胞样 (ABC) DLBCL 和无法分类亚型,标准化疗后 GCB 型结局更优(图3A)。基因表达谱可确定具有不同特征的DLBCL亚型,类似于COO。随后建立免疫组化 Hans 分类器,根据CD10、BCL6和 IRF4/MUM1 表达区分 GCB 和 non-GCB 亚型。免疫组化仍是临床常规中鉴定 DLBCL 起源细胞最广泛使用的方法,在预测基因表达谱亚型或预后分子学亚组方面的一致性约为80%。

通过荧光原位杂交,大约10-15%的新诊断DLBCL 患者携带MYC 基因重排。同时具有 MYC和BCL2 重排的患者被ICC 和 WHO-2022称为高级别 B 细胞淋巴瘤,标准免疫化疗下预后不佳。双打击淋巴瘤通常在 GCB 型 DLBCL 中检测到(图3A)。MYC 和 BCL6 易位是 ICC 中高级别 B 细胞淋巴瘤的临时类别,但WHO-2022 分类中并非如此。BCL2 和 MYC 蛋白过表达但没有相应基因易位的淋巴瘤称为双表达淋巴瘤,发生率在诊断时约30%,通常为活化B细胞起源,预后居中(图3A)。研究已经确定了高级别淋巴瘤的特异性基因表达特征,特别是 MYC 和 BCL2(BCL6 易位并无),可能有助于其鉴别诊断。
全外显子测序和拷贝数畸变等深度遗传学分析的使用,进一步改善了 DLBCL 的分子特征(图3B)。不同的研究组已确定相似的分子亚群:Schmitz等最初定义了与一线免疫化疗不同反应相关的 DLBCL 的4种基因亚型:包括预后不良的MCD(同时发生 MYD88L267P和 CD79B 突变)和N1(NOTCH1突变)亚型,以及预后良好的BN2(BCL6融合和 NOTCH2 突变)和EZB(EZH2和 BCL2 易位)亚型。Chapuy 等报告了5个 DLBCL 亚群 (C1–C5):低危DLBCL(C1,与 MYD88[非 L265P] 突变及 NOTCH 和 BCL6 信号转导干扰相关)、高危ABC-DLBCL(C5,MYD88[L265P] 突变)、具有不同结局和靶病变的 GCB-DLBCL 的2个亚群(C3,BCL2、PI3K、EZH2和CREBBP;C4,JAK/STAT 和BRAF/MEK1),以及 TP53 双等位基因失活、CDKN2A缺失和相关基因组不稳定的 ABC-GCB 非依赖性组(C2)。LymphGen 算法定义的 DLBCL 分类法进一步扩展了这些亚组,并提供了这两项研究的统一视图(图3B)。该分类将 DLBCL 分为7种基因亚型,具有不同的致癌通路参与、基因表达表型、肿瘤微环境、生存率和潜在治疗靶点。然而,这些基因亚型尚未在常规临床实践中诊断应用,即使采用这些复杂的方法,仍有相当比例(约40%)的 DLBCL 未得到分配。
DLBCL 通常对初始治疗反应非常好,三分之二的患者可治愈。在前24个月内未复发的患者预后非常好,预期寿命与一般人群相似,但有时可观察到晚期复发。
对于当前利妥昔单抗治疗时代的临床风险分层,NCCN-IPI在区分低危和高危(5年总生存率<50%)DLBCL患者方面优于IPI。其改进了年龄、乳酸脱氢酶和特定结外部位受累,分为低危(0-1分)、低-中危(2-3分)、高-中危(4-5分)和高危(≥6分)。
对于大多数晚期 DLBCL 患者,R-CHOP 3周/周期共6个周期是标准治疗,而14天/周期的剂量强化未证明更优。在 Alliance/CALGB 50303 3 期组间研究中,剂量强化方案DA-EPOCH-R(剂量调整的依托泊苷、泼尼松、长春新碱、环磷酰胺、多柔比星和利妥昔单抗),与 R-CHOP 相比毒性更大,但未显著改善2年无进展生存期或总生存期。在 FLYER 研究中,局限期DLBCL(I–II期、非大包块、乳酸脱氢酶正常和 ECOG 体能状态为0-1分)仅接受4个周期 R-CHOP 治疗后再接受2个周期利妥昔单抗单药治疗即可充分治疗,其3年无进展生存期与6个周期 R-CHOP 治疗相似。然而,在首次报道 R-CHOP 后近20年,POLARIX研究是首个成功挑战该化疗标准的3期研究,用维泊妥珠单抗(Polatuzumab vedotin)代替长春新碱并联合 R-CHP 治疗。与 R-CHOP 组相比,该改良方案显著改善2年无进展生存期(76.7% vs 70.2%;p=0.02),而两组的总生存期保持相似。亚组分析提示明显获益,尤其是对于高危患者(年龄<60岁、IPI 3-5或 ABC 亚型)。维泊妥珠单抗联合R-CHP于2023年4月获得FDA批准,用于DLBCL的一线治疗。
使用 PET-CT 监测治疗结束时反应,PET阳性残留结合 Deauville 评分5表明治疗失败;这些患者可能需要全身挽救治疗。在 Deauville 评分为4的情况下,应考虑再次活检,挽救治疗或严格控制是适当选择。
很多研究致力于改善一线治疗,包括具有病理学高危生物学特征(non-GCB 亚型、双打击淋巴瘤)的 DLBCL 或根据不良临床特征预计对 R-CHOP 无应答的患者。
在non-GCB DLBCL 患者中比较伊布替尼+R-CHOP与 R-CHOP 治疗(PHOENIX研究),结果显示毒性作用增加导致 R-CHOP 给药受到影响,故未改善无事件生存期、无进展生存期或总生存期。另一项 III 期研究 (ROBUST) 在non-GCB DLBCL 患者中评估在标准 R-CHOP 基础上加用来那度胺,结果显示无进展生存期略有改善,但总生存期无改善。在既往未经治疗的non-GCB DLBCL 患者中,硼替佐米联合 R-CHOP 治疗与 R-CHOP 单药治疗相比也未能显著改善结局。
对于伴MYC 和BCL2、BCL6或两者重排的高级别 B 细胞淋巴瘤,DA-EPOCH-R可能是诱导持久缓解的首选免疫化疗方案。对于这种侵袭性 DLBCL 亚型,多中心、开放标签、单臂 ZUMA-12 研究评估了 CD19 CAR-T 细胞产品 axicel 作为高危 DLBCL 患者一线治疗的疗效,完全缓解率为78%。
在新诊断 DLBCL 患者中使用 CNS 预防越来越受到质疑。两项大型回顾性研究分析了 CNS 预防的最佳时机和应用,尽管其目的并非评价 CNS 预防的一般效果,但与历史 CNS 复发率相比,它们未显示复发率的获益。然而,作者认为大剂量甲氨蝶呤预防足以用于睾丸和肾脏受累的超高危患者。如有指征,治疗结束时应给予大剂量甲氨蝶呤,至少2个周期。推荐的靶向治疗总结见表2。
不久之前,挽救化疗序贯ASCT还是适合移植的复发或难治性 DLBCL 患者的标准治疗。对于不适合移植的患者,几种获批的替代治疗选择显示有效:维泊妥珠单抗联合利妥昔单抗和苯达莫司汀与利妥昔单抗联合苯达莫司汀相比,完全缓解率显著更高,死亡风险降低58%。抗 CD19 单克隆抗体 tafasitamab 联合来那度胺可使高比例的不适合移植患者达到完全缓解。FDA 目前批准了3种自体 CAR-T 细胞产品用于治疗既往接受过≥2线全身治疗的复发或难治性 DLBCL 患者:axicabtagene ciloleucel(axicel)、lisocabtagene maraleucel(lisocel) 和tisagenlecleucel(tisacel)。近期在适合移植的早期复发患者(R-CHOP治疗完成后12个月内)中比较了三款CAR-T产品与当前标准治疗(挽救治疗序贯大剂量化疗和 ASCT)。接受 axicel 治疗的患者的无事件生存率显著改善(2年后41% vs 16%;p<0.001),缓解率也显著高于标准治疗。同样,与标准治疗相比,lisocel还显著改善了无事件生存率(12个月后44.5% vs 23.7%;p<0.0001),并提高了缓解率。而与 ASCT 相比,tisacel未获得任何获益;但该结果可能是由于临床试验设计和 CAR-T 细胞产品的延迟可用性,而非由于产品活性不同。由于2/3项研究显示 CAR-T 细胞产品在首次复发中具有优效性,该治疗策略成为适合移植的早期首次复发 DLBCL 患者的新标准治疗。推荐的靶向治疗总结见表2。
新型免疫治疗替代方案目前挑战了复发或难治性 DLBCL 的治疗方案。双特异性 T 细胞衔接系统治疗,如靶向 CD20 和 CD3 的抗体,也在研究中。使用递增剂量以减轻首次输注期间发生的副作用,mosunetuzumab、epcoritamab(皮下)以及格菲妥单抗(具有两个 CD20 结合基序的二比一形式,奥妥珠单抗预处理)作为单药均显示令人鼓舞的结果,正在与其他药物联合评价。格菲妥单抗目前已在加拿大和美国获批,epcoritamab目前已在美国获批用于三线或三线以后的治疗。抗体-药物偶联物为复发或难治性 DLBCL 提供了另一种有前景的替代治疗方法,其通过与靶向肿瘤细胞表面抗原的单克隆抗体偶联,选择性地将细胞毒性药物递送至肿瘤细胞。Loncastuximab tesirine 是一种与吡咯苯二氮卓二聚体偶联的抗 CD19 抗体,2期 LOTIS-2 研究中评价证实对既往接受过多线治疗的复发或难治性 DLBCL 患者有效。
Burkitt淋巴瘤
Burkitt 淋巴瘤是一种生发中心起源的高度侵袭性 B 细胞淋巴瘤,可分为为地方型、散发型与免疫缺陷相关型。B 细胞通常表达CD45、CD20、CD79a、CD10和BCL6,BCL2阴性,增殖率极高(Ki67指数高达100%)。Burkitt 淋巴瘤的遗传学特征为t(8;14)染色体易位导致 MYC 组成性激活。在40–70%的t(8;14) 易位病例中,其他突变包括转录因子 TCF3 或其阴性抑制剂ID3,以及 CCND3 和 TP53 或 CDKN2A 失活改变。Burkitt淋巴瘤的基因表达谱显示高表达MYC 靶标和生发中心相关 B 细胞基因,可能有助于更可靠地区分其与 DLBCL。
考虑到 Burkitt 淋巴瘤的快速倍增时间,患者最常表现为大肿瘤包块、骨髓受累、高血清乳酸脱氢酶水平,有时可出现自发性肿瘤溶解综合征的体征。由于该淋巴瘤亚型倾向于播散至CNS,因此应在初诊时进行腰椎穿刺分期。
立即开始治疗非常重要,应降低肿瘤溶解风险,并应包括 CNS 预防。强化免疫化疗方案,如环磷酰胺、长春新碱、多柔比星、甲氨蝶呤、异环磷酰胺、依托泊苷和阿糖胞苷 (CODOX-M/IVAC) 可获得极佳缓解率。在短期强化化疗方案中加入利妥昔单抗可改善 Burkitt 白血病或淋巴瘤成人患者的无事件生存期和总生存期。在散发型或免疫缺陷相关型 Burkitt 淋巴瘤成人中,输注方案 DAEPOCH 联合利妥昔单抗高效且毒性特征可耐受。一项在新诊断高危 Burkitt 淋巴瘤患者中比较 R-CODOX-M/IVAC 与 DA-EPOCH-R 的欧洲3期研究启动后,由于招募水平较低而不得不提前关闭(纳入89例患者)。中位随访19.1个月,生存率未见明显差异。但对于 CNS 受累患者,作者认为 DA-EPOCH-R 存在不足。
总结
在过去十年中,我们对 B 细胞淋巴瘤生物学基础的理解有了很大的提高,为这组异质性疾病的诊断、预后和治疗管理的众多新发展铺平了道路。考虑到遗传和其他分子数据在淋巴肿瘤评价中的重要性越来越大,这种理解的增加导致修订了 B-NHL 分类。深度遗传学分析的使用导致定义了高危疾病的预后相关遗传特征。然而,除了套细胞淋巴瘤中的 TP53 突变,这些预后评分还没有进入诊断常规,仍需要改善其临床适用性。
目前的研究仍在继续评估 DLBCL 分子特征亚群的靶向治疗。在惰性淋巴瘤中,基于化疗的一线方法越来越受到靶向治疗的挑战。最后,免疫治疗方法,主要是过继性 T 细胞治疗,正在成为多种淋巴瘤亚型疾病复发的标准治疗,包括DLBCL(CAR-T细胞)及惰性淋巴瘤(双特异性抗体)。这些知识可能导致更多基于风险和个体化的治疗策略,从而使患者受益。
参考文献
Elisabeth Silkenstedt, Gilles Salles, Elias Campo, Martin Dreyling.B-cell non-Hodgkin lymphomas.Lancet . 2024 Apr 10:S0140-6736(23)02705-8. doi: 10.1016/S0140-6736(23)02705-8.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#B细胞非霍奇金淋巴瘤# #B-NHL#
38