X连锁无丙种球蛋白血症:症状与体征、病因、流行病学、诊断与治疗
2022-08-27 MedSci原创 MedSci原创

X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)为X-连锁隐性遗传病,是由于人类B细胞系列发育障碍引起的原发性免疫缺陷病。本病仅见于男性,又名Bruton。2
X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)为X-连锁隐性遗传病,是由于人类B细胞系列发育障碍引起的原发性免疫缺陷病。本病仅见于男性,又名Bruton。2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,X-连锁无丙种球蛋白血症被收录其中。
一、一般概述
无丙种球蛋白血症是一组遗传性免疫缺陷,其特征是由于血液和淋巴液中缺乏特定的淋巴细胞,导致血液中抗体浓度低。抗体是蛋白质(免疫球蛋白、(IgM)、(IgG)等),它们是免疫系统的关键和关键成分。如果免疫系统要完成对抗细菌、病毒和其他威胁身体的外来物质的工作,它们是必不可少的。产生丙种球蛋白的特化前体细胞不能正常发育或发挥功能,导致称为 B 细胞的成熟淋巴细胞数量不足。
无丙种球蛋白血症的类型有:X 连锁无丙种球蛋白血症 (XLA)、更罕见的 X 连锁无丙种球蛋白血症伴生长激素缺乏症(报告约 10 例)和常染色体隐性遗传性无丙种球蛋白血症 (ARAG)。所有这些疾病的特点是免疫系统减弱,必须通过给予丙种球蛋白来加强免疫系统以抵抗感染。
二、症状与体征
无丙种球蛋白血症的主要症状是由于 B 淋巴细胞缺陷导致特异性免疫反应失败导致的一系列细菌感染。这些淋巴细胞控制抗体的产生。患有 X 连锁原发性无丙种球蛋白血症的男性通常仅在出生后第一年的晚期才开始出现此类感染的迹象,此时来自母亲的 IgG 抗体已经耗尽。
几乎任何肠道病毒家族和脊髓灰质炎病毒的感染都可能导致患有无丙种球蛋白血症的儿童出现异常严重的疾病。回声病毒感染可引起一组与皮肌炎非常相似的症状。这些症状可能包括肌肉无力,通常在臀部和肩部区域,以及吞咽困难。眼睛、指关节和肘部周围可能会出现斑片状、微红色的皮肤区域,偶尔会出现在膝盖和脚踝上。
支原体细菌引起的感染可导致患有原发性无丙种球蛋白血症的儿童出现严重的关节炎,包括关节肿胀和疼痛。流感嗜血杆菌是最常见的产生粘液的感染(化脓性),发生在 X 连锁无丙种球蛋白血症患者中。儿童也可能反复感染肺炎球菌、链球菌和葡萄球菌,很少感染假单胞菌。
患有 X 连锁无丙种球蛋白血症的男性血液中循环的 IgA、IgG 和 IgM 抗体水平非常低。专门的白细胞(中性粒细胞)破坏细菌、病毒或其他入侵生物(微生物)的能力受损。这是因为中性粒细胞需要来自免疫系统的抗体才能开始破坏入侵的细菌(调理作用)。患有无丙种球蛋白血症的儿童的循环中性粒细胞水平可能持续偏低,或者在患有这些疾病的人中可能会起起伏伏(周期性、短暂性中性粒细胞减少症)。 X连锁无丙种球蛋白血症患儿的B淋巴细胞数量少于正常数量的百分之一。
5 或 6 个家庭中只有大约 10 人被诊断出患有 X 连锁无丙种球蛋白血症伴生长激素缺乏症。这些家庭中的男孩的 B 淋巴细胞数量减少或检测不到。临床医生和遗传学家推测 BTK 基因中的第二个突变,非常接近导致 XLA 的该基因中的突变,是导致无丙种球蛋白血症和身材矮小的组合的原因。
据报道,常染色体隐性遗传的无丙种球蛋白血症是由影响 B 细胞发育的基因引起的。
另外,XLA 患儿容易出现中耳炎、慢性鼻窦炎、营养不良、贫血、粒细胞减少、血小板减少、生长激素缺乏症及甲状腺激素紊乱等并发症状。XLA 患儿还可发生肿瘤、自身免疫和炎症性疾病,包括类风湿性关节炎、皮肌炎等。据报道,患儿的感音神经性听力损失和湿疹的发生率也较高。
三、病因
X连锁无丙种球蛋白血症(B淋巴细胞缺陷)作为X连锁隐性遗传特征遗传。名为BTK的异常基因已被定位到基因位点Xq21.3-q22。 BTK 基因的不同突变导致 X 连锁无丙种球蛋白血症伴生长激素缺乏症。 ARAG 的遗传原因要复杂得多,涉及已映射到不同染色体上的基因座的其他基因:22q11.21、14q32.33 和 9q34.13。三个位点的基因分别称为 IGLL1、IGHM 和 LCRR8。
遗传疾病是由来自父亲和母亲的染色体上特定性状的基因组合决定的。
X连锁遗传疾病是由X染色体上的异常基因引起的疾病,主要发生在男性身上。在其中一条 X 染色体上存在疾病基因的女性是该疾病的携带者。携带者女性通常不会表现出症状。
患有 X 连锁疾病的男性会将疾病基因传给他们所有将成为携带者的女儿。男性不能将 X 连锁基因传递给他的儿子,因为男性总是将 Y 染色体而不是 X 染色体传递给男性后代。
隐性遗传疾病发生在个体遗传了同一性状的异常基因的两个拷贝时,每个拷贝来自父母一方。如果一个人接受一个正常基因和一个疾病基因,该人将成为该疾病的携带者,但通常不会出现症状。每次怀孕,两个携带者父母都通过缺陷基因并有受影响的孩子的风险是 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。一个孩子从父母双方那里获得正常基因并在该特定特征上遗传正常的机会是 25%。男性和女性的风险相同。
所有个体都携带 4-5 个异常基因。近亲(近亲)的父母比无关父母携带相同异常基因的可能性更高,这增加了患隐性遗传疾病的孩子的风险。
四、流行病学
原发性无丙种球蛋白血症是一种罕见的疾病,几乎只发生在男性身上,尽管一些女性受到某些类型的这种疾病的影响。
美国 XLA 患者登记处数据显示 379 000 例活产婴儿中约有 1 例(190 000 名男性中有 1 例)。我国目前没有关于 XLA 发病率及患病率的相关数据。
五、鉴别诊断
以下疾病的症状可能与原发性无丙种球蛋白血症相似。比较可能有助于鉴别诊断:
常见可变免疫缺陷 (CVID) 是一种罕见的免疫缺陷疾病,其特征是肺部、鼻窦或耳朵的反复感染。与 CVI 相关的症状和发现的范围和严重程度可能因病例而异。在某些情况下,患有 CVID 的人更容易发生胃肠系统感染,并且可能患某些类型的癌症(例如非霍奇金淋巴瘤和胃癌)的风险更高。此外,一些患有 CVID 的人患有自身免疫性疾病,如免疫性血小板减少性紫癜,会导致异常瘀伤和出血。 CVI 的症状通常在生命的第二个到第四个十年变得明显。 CVID 被认为是由与 B 细胞产生相关的基因突变引起的,这些 B 细胞产生针对感染因子的抗体。在大多数情况下,CVID 可能是由遗传和环境因素共同引起的,但在一些家庭中已经描述了常染色体隐性遗传和常染色体显性遗传。

高 IgM 综合征 (HIGM) 是一种罕见的原发性免疫缺陷疾病,通常作为 X 连锁隐性遗传病。患有这种疾病的人 IgG、IgA 和 IgE 抗体水平较低。 IgM 抗体水平可能较高或在正常范围内。症状和身体检查结果通常在生命的第一年或第二年变得明显。 HIGM 的特征是中耳、鼻窦、肺、眼睑和眼睛白色部分的膜、皮肤和/或其他区域的反复细菌感染。受影响的儿童可能会出现营养吸收障碍、慢性腹泻和体重增加失败(发育迟缓)以及扁桃体肿大和/或肝脏和脾脏肿大(肝脾肿大)。此外,受影响的个体容易发生血液自身免疫性疾病,例如中性粒细胞减少症,其中某些白细胞水平降低。因为大约 70% 的 HIGM 报告病例是 X 连锁的,所以绝大多数受影响的个体是男性。然而,也描述了该疾病的常染色体隐性和常染色体显性形式。
严重联合免疫缺陷(SCID)是原发性免疫缺陷病中最严重的一种。患有 SCID 的人会反复感染,因为 B 淋巴细胞和 T 淋巴细胞都没有足够数量,或者它们出现故障。如果不治疗,这种疾病可能会导致频繁、严重的感染、生长迟缓,并可能危及生命。这种疾病的其他症状可能包括体重减轻、虚弱、中耳感染和皮肤感染。
WAS 相关疾病是由 WAS 基因突变引起的影响免疫系统的一系列疾病。这些疾病包括 Wiskott-Aldrich 综合征、X 连锁血小板减少症和 X 连锁先天性中性粒细胞减少症。 WAS 基因异常导致 WASP 蛋白缺乏,从而导致血小板计数低(血小板减少症)。 WAS 相关疾病通常出现在婴儿期,其特征是血性腹泻、反复感染、脱屑、瘙痒、皮疹(湿疹)和皮肤上出现紫色小斑点(瘀点)。卡氏肺孢子虫肺炎 (PCP) 和颅内出血的发展可能是早期的、危及生命的并发症。后来的潜在并发症包括红细胞破坏(溶血性贫血)、关节炎、血管炎、肾和肝损伤。受影响的个体患淋巴瘤的风险增加,尤其是在接触 Epstein-Barr 病毒后。与 WAS 相关的疾病变化很大,即使在同一家庭中的个体中也是如此。
IgA 缺乏症是一种与无丙种球蛋白血症相关的抗体缺乏症,其特征是血液中 IgA 水平低,而 IgG 和 IgM 水平正常或升高。 IgA 缺乏是最常见的原发性免疫缺陷。免疫球蛋白同位素的其他缺陷是 IgM 缺陷和 IgG 亚类缺陷。
补体成分 3 缺乏症是一种罕见的遗传性免疫缺陷,其特征是反复呼吸道感染、皮肤感染、反复中耳感染和鼻窦炎。 这种疾病的症状与一些无丙种球蛋白血症的症状非常相似。 其他症状可能包括肺炎、血液细菌感染(败血症)和/或脑膜炎症(脑膜炎)。 其他疾病也可能与补体成分 3 缺乏有关,包括血管炎症(血管炎)、关节痛(关节痛)和自身免疫性疾病如狼疮(系统性红斑狼疮)。
婴儿期暂时性低丙种球蛋白血症,即血清IgG很低,而IgA和IgM正常。外周血中B细胞计数正常。淋巴结活检虽缺少成熟浆细胞,但有浆细胞样淋巴细胞。一般不超过18个月即可恢复合成免疫球蛋白的能力。
六、诊断
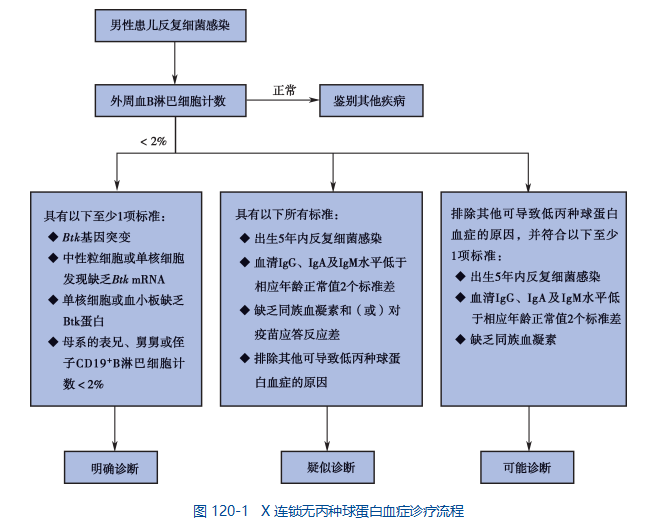
根据临床表现和实验室结果如血清免疫球蛋白减少,特异性抗体反应缺乏,外周血成熟 B 细胞缺如等,不难对 XLA 做出临床拟诊,确诊有赖于 Btk 基因检测。
1. XLA 临床诊断依据
(1) 男性;
(2) 反复较严重细菌感染(呼吸道、胃肠道、皮肤及其他深部感染),抗生素治疗效果不佳;
(3) 伴或不伴有自身免疫性疾病;
(4) 伴或不伴有母系家族中类似疾病表现的男性患者。
2. XLA 的实验室诊断标准 目前采用的是 1999 年泛美免疫缺陷工作组和欧洲免疫学会制定的标准(表 120-1)。

七、治疗
免疫球蛋白替代疗法可控制大多数 XLA 患儿的感染症状,全身状况可迅速改善。目前有静脉途径(IVIG) 和皮下途径两种(SIG) 两种, 国内主要采用 IVIG。IVIG 治疗XLA 的总原则是:早用比晚用效果好;较大剂量比小剂量好。如果 IVIG 治疗开始较晚, 感染所致的器质性损害将是不可逆的。临床中常用剂量为 400~600mg/kg,每 3~4 周 1 次。血清 IgG 维持 5g/L 以上,感染明显减少,但有研究表明,此浓度不能提供足够保护,部分患者可能需更高剂量的 IgG 以控制感染,IVIG 用量应个体化。对于有明确感染的 XLA 患者应积极应用抗生素,根据药敏结果及时调整抗生素的种类及疗程。但关于在 XLA 患者中使用预防性抗生素相关问题,目前尚未达成共识。
除 IVIG 替代性治疗外,尚需各种支持疗法,包括营养、生活及卫生条件的改善,预防感染,适当进行体育锻炼,保持良好心理状态,防治各种并发症等。对于明确诊断 XLA 的患者,禁止口服脊髓灰质炎疫苗。
目前,人们认为异基因造血干细胞移植对 XLA 的风险大于益处,用于纠正自体造血干细胞的基因疗法仍在研究中,迄今尚未开始对人类的临床试验。
当发生细菌感染时,会为患有无丙种球蛋白血症的人开具抗生素。一些患者接受抗生素治疗作为预防措施(预防性)。应尽可能保护所有免疫缺陷的人免于接触传染病。应尽可能避免使用皮质类固醇或任何抑制免疫系统的药物(免疫抑制剂),以及可能损害脾脏的粗暴接触运动等体育活动。
在 IgM 升高的免疫缺陷患者中,存在过度出血的倾向,这与血液中异常低水平的循环血小板(血小板减少症)有关。这可能会使任何外科手术复杂化。
建议对无丙种球蛋白血症患者及其家人进行遗传咨询。其他治疗是对症治疗和支持治疗。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考文献:
殷勇,袁姝华 . 儿童 X 连锁无丙种球蛋白血症 . 中华实用儿科临床杂志,2018,33(4):288-291.
https://www.chard.org.cn/#/knowledge/jbzsk/detail/153
Kumar A, Teuber SS, Gershwin ME. Current perspectives on primary immunodifieciency diseases. Clin Dev Immunol. 2006;13:223-59.
Stangel M, Pul R. Basic principles of intravenous immunoglobulin (IVIg) treatment. J Neurol. 2006;253 Suppl 5:v18-v24.
Winkelstein JA, Marino MC, Lederman HM et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore). 2006;85:193-202.
Rose ME, Lang DM. Evaluating and managing hypogammaglobulinemia. Cleve Clin J Med. 2006;73:133-37, 140, 143-44.
Lawrence T, Puel A, Reichenbach j, et al. Autosomal-dominant primary immunodeficiencies. Curr Opin Hematol. 2005;12:22-30.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Bruton agammaglobulinemia tyrosine kinase: BTK. Entry No: 300300; Last Update:10/11/2006.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Agammaglobulinemia, X-Linked. Entry No: 300755; Last Update:12/19/08.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Agammaglobulinemia, Non-Bruton Type, Autosomal recessive. Entry No. 601495; Last Update: 1/6/06.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Hypogammaglobulinemia and isolated growth hormone deficiency, X-linked. Entry No. 307200; Last Update: 9/27/01.
Conley ME and Howard VC. Updated 12/21/05. X-Linked Agammaglobulinemia. In: GeneReviews at Genetests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2009. Available at http://www.genetests.org. Accessed 1/09.
Little FF. Agammaglobulinemia. Medical Encyclopedia. MedlinePlus. Update date 6/21/2006. 3pp.
www.nlm.nih.gov/medlineplus/ency/article/001307.htm
Accessed on 1/04/2007
X-linked Agammaglobulinemia. Children’s Hospital Boston. ©2006. 3pp.
www.childrenshospital.org/az/Site1803/mainpageS1803P0.html
Accessed on 1/04/2007
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#X连锁#
114
#球蛋白#
69
#诊断与治疗#
92
#丙种球蛋白#
99
#流行病#
93