
Nature Microbiology:单细胞大规模并行多路微生物测序识别罕见的细菌种群和描述噬菌体感染
2023-10-05 Jenny Ou MedSci原创 发表于上海

本文将M3-seq应用于数十万个细胞,揭示了罕见的种群以及与压力反应和噬菌体感染特征相关的赌注对冲的见解。
细菌具有适应多样化和不断变化的环境的非凡能力。面对不可预测的环境压力,允许人口蓬勃发展的一个策略是单个细胞的专业化。这些专业化可以表现为形态变化(例如,革兰氏阳性生物的孢子)或视觉上无法区分但功能不同的状态(例如,金黄色葡萄球菌和大肠杆菌中罕见的抗生素耐药性“瘀斑”表型)。

研究此类专业化的一个有前途的方法是测量单个细胞如何协调基因表达。对于哺乳动物细胞,这种测量是通过单细胞RNA测序(scRNA-seq)实现的。尽管开创性地努力开发类似的细菌工具,但目前研究微生物的技术却落在了后面。现有的细菌scRNA-seq方法包括MATQ-seq、PETRI-seq、microSPLiT、par-SeqFISH和ProBac-seq。这些方法中的每一种方法都使用不同的策略来索引细胞及其转录物,每种方法都有优点和缺点。
MATQ-seq将单个细胞分离到多孔板的单独井中,并执行单独的索引反应以生成测序库。这种“索引”方案在规模上本质上是有限的。相比之下,其余每种方法都允许在单个实验中跨细胞池对单细胞基因表达进行表征,通过原位探针杂交(SeqFISH和ProBac-seq)或分裂池组合索引(PETRI-seq,microSPLiT)实现多路转录检测。这些方法在细菌中建立了单细胞转录组学领域,但缺点仍然存在。
基于杂交的方法依赖于预先设计的物种和基因特异性探针,从而限制了无偏见的发现,而组合索引平台具有来自核糖体(r)RNA的大量信号,这可能会影响信使(m)RNA的检测。鉴于这些考虑,2023年8月31日发表在Nature Microbiology的文章,开发了大规模并行、多路复用微生物测序(M3-seq),这是一种细菌中scRNA-seq的方法,它结合了基于板的原位索引与基于液滴的索引和后rRNA耗尽。
在本文的研究的同时,还报告了另一种基于液滴的scRNA-seq方法,称为BacDrop。这种方法在原位执行rRNA耗尽,而M3-seq在库扩增后执行rRNA耗尽,从而降低失去未扩增的非rRNA转录物的风险,并可能提高灵敏度。M3-seq能够以敏感的mRNA捕获在许多样本中对单个细菌细胞进行大规模并行基因表达分析。通过将M3-seq应用于数十万个细胞,研究人员揭示了枯草芽孢杆菌的独立噬菌体诱导程序,大肠杆菌的对冲亚群以及噬菌体感染的详细异质性。
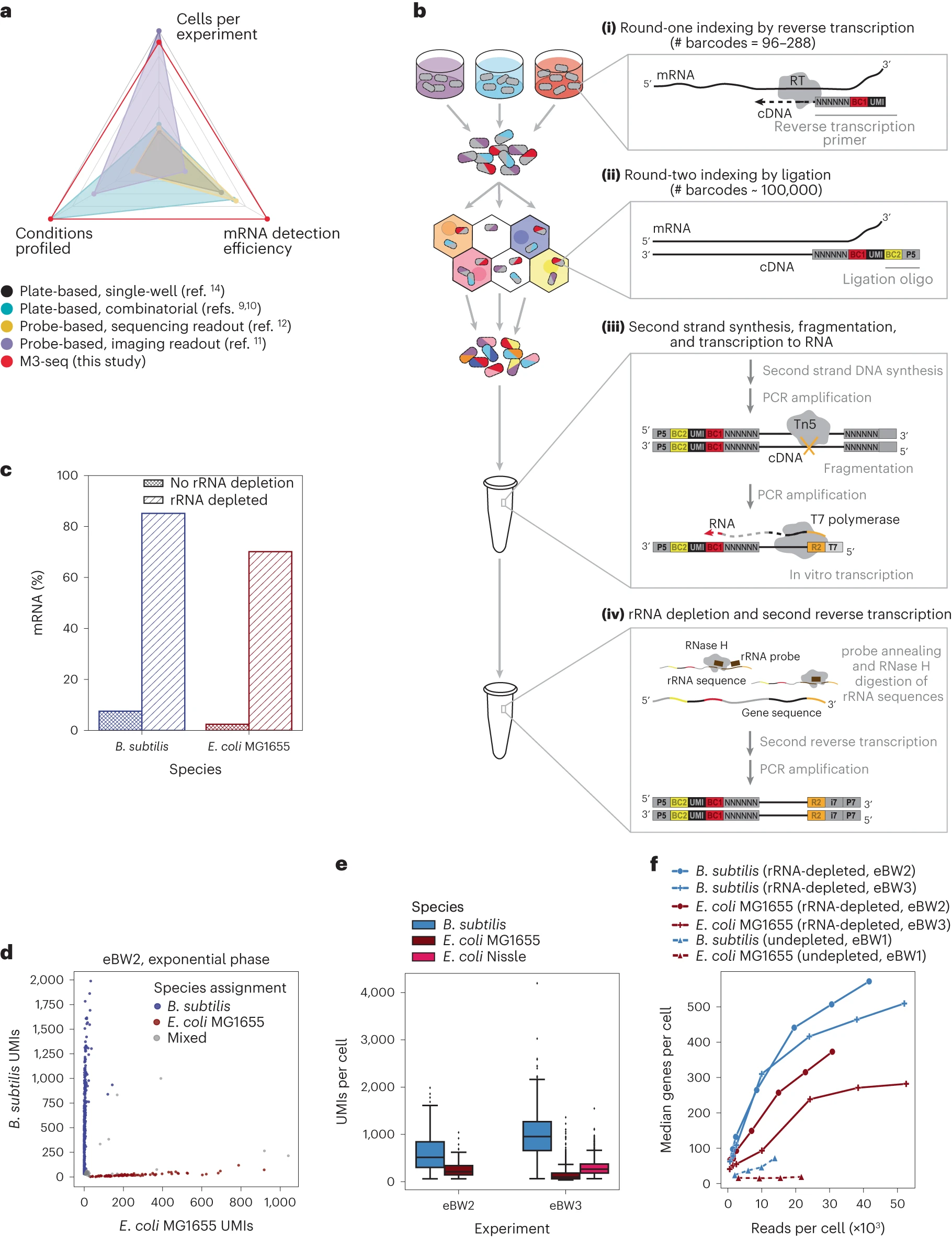
开发M3-seq平台,用于具有后rRNA耗尽的单细胞RNA测序
研究人员开发了大规模并行、多路复用微生物测序(M3-seq)——一个用于细菌的单细胞RNA测序平台,将组合细胞索引与后rRNA耗尽配对。研究结果表明,在单个实验中,M3-seq可以在一系列条件下剖析来自不同物种的细菌细胞。然后,我们将M3-seq应用于数十万个细胞,揭示了罕见的种群以及与压力反应和噬菌体感染特征相关的赌注对冲的见解。
综上所述,本文研究看到了多个生物系统,本文的技术已经成熟,可以应用这些系统。毫无疑问,一个关键应用将是宿主-病原体相互作用,例如,揭示细菌如何调动噬菌体免疫机制。此外,这种应用不必局限于细菌细胞。由于使用随机引物和rRNA耗尽方案的普遍性,本文的方法也可以用于研究哺乳动物细胞如何对细胞内病原体的感染做出反应,以及这些感染病原体如何对宿主因素做出反应。
原文出处
Wang, B., Lin, A.E., Yuan, J. et al. Single-cell massively-parallel multiplexed microbial sequencing (M3-seq) identifies rare bacterial populations and profiles phage infection.Nat Microbiol 8, 1846–1862 (2023). https://doi.org/10.1038/s41564-023-01462-3
 s41564-023-01462-3.pdf
s41564-023-01462-3.pdf
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




看来菌群在临床研究中还是大有可为
79