
JAHA:小儿扩张型心肌病基因型与心脏结局
2021-12-23 MedSci原创 MedSci原创
肌节变异在儿科DCM患者中很常见。该研究证明了诊断年龄与心脏结局之间存在基因型特异性关联。特别是,MYH7与伴有左心室致密化不全特征的DCM患者具有结构域特异性关联。
小儿扩张型心肌病(DCM)是一个众所周知的临床疾病;然而,DCM的表型-基因型相关性尚未完全明确。

近日,心血管疾病领域权威杂志JAHA上发表了一篇研究文章,在这项研究中,研究人员旨在明确儿科DCM先证者中提供与危及生命的心脏结局的基因型关联。

研究人员在一家大型儿科转诊中心(2007-2016年)对DCM儿童进行了回顾性研究,排除了综合征、化疗引起的和先天性心脏病所致的DCM。遗传变异由专家小组和独立的临床实验室来进行判定。
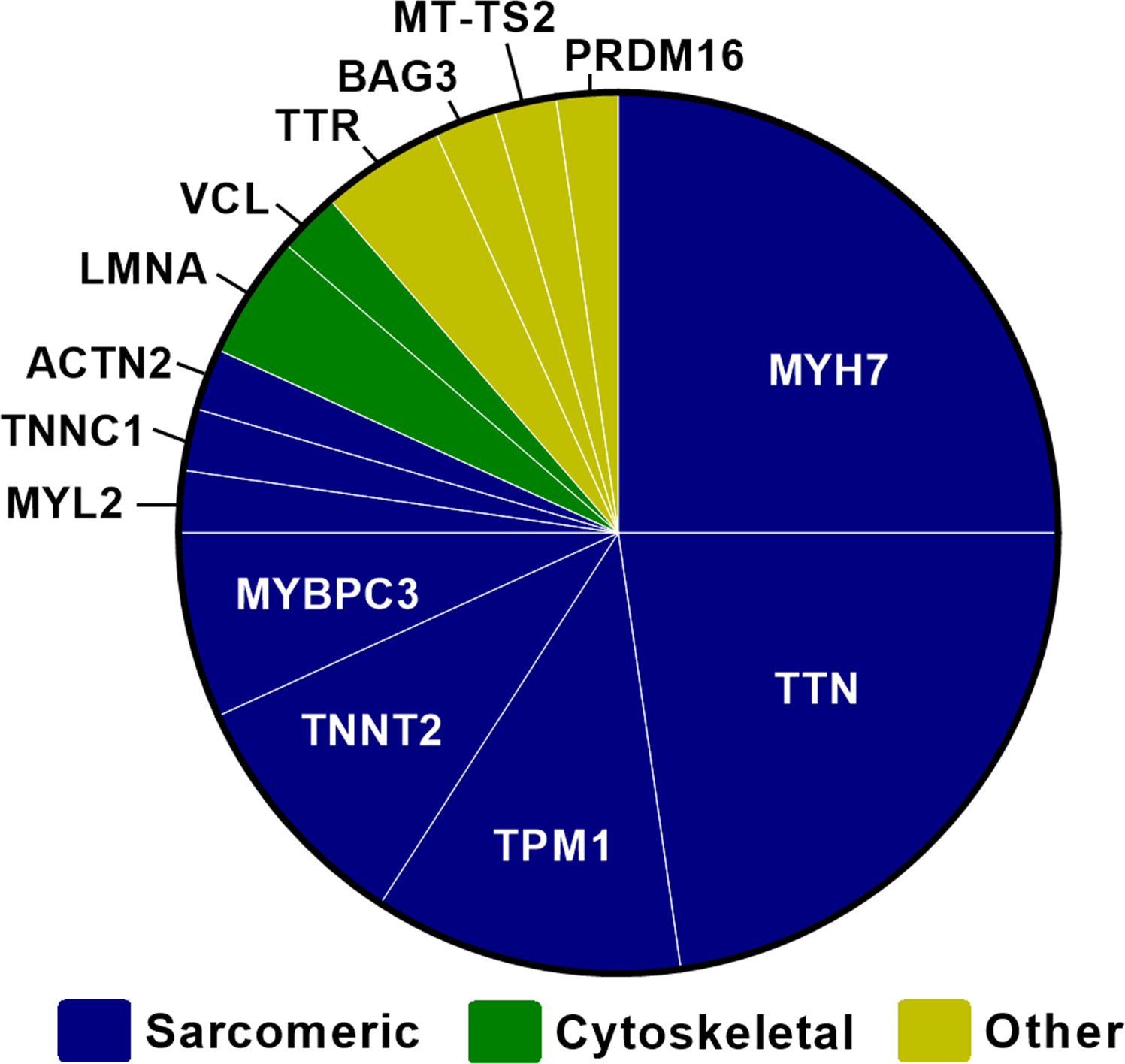
在109名DCM儿童病例的队列中,诊断时的平均年龄为4.2岁(SD为5.9),47%的DCM儿童发生危及生命的心脏结局(42%为心脏移植,5%为死亡病例)。40人/109名(37%)受试者存在一种或多种致病性/可能致病性突变,36人/44名(82%)受试者的致病性/可能致病性突变发生在肌节基因。家族性心肌病患者的致病性/可能致病性突变的频率没有差异(15人/33名受试者有家族史 vs. 25人/76名受试者没有家族史,P=0.21)。在青少年期诊断DCM的儿童中TTN截断突变的发生比例较高(26%的青少年 vs. 6%的年幼儿童,P=0.01),但具有这些TTN变异的婴儿和青少年都发生了危及生命的心脏结局。伴有左心室致密化不全特征的6名DCM患者的突变全部发生在1和600氨基酸之间的MYH7基因。
由此可见,肌节变异在儿科DCM患者中很常见。该研究证明了诊断年龄与心脏结局之间存在基因型特异性关联。特别是,MYH7与伴有左心室致密化不全特征的DCM患者具有结构域特异性关联。家族史并不能预测致病性/可能的致病性变异,这强调了应在所有特发性DCM儿童中考虑进行基因检测。
原始出处:
Rabia S. Khan,et al.Genotype and Cardiac Outcomes in Pediatric Dilated Cardiomyopathy.JAHA.2021.https://www.ahajournals.org/doi/full/10.1161/JAHA.121.022854
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#基因型#
57
#心肌病基因#
0
#扩张#
63
肌节变异在儿科DCM患者中很常见。该研究证明了诊断年龄与心脏结局之间存在基因型特异性关联。特别是,MYH7与伴有左心室致密化不全特征的DCM患者具有结构域特异性关联。家族史并不能预测致病性/可能的致病性变异,这强调了应在所有特发性DCM儿童中考虑进行基因检测。
68
#心肌病##扩张性心肌病#
117
#AHA#
43
#肌病#
58