
39岁男性,右上肢无力1年半,加重4个月,体检发现右上肢呈上运动神经元病损,合并感觉障碍。肌萎缩侧索硬化?脊肌萎缩症?颈椎病?还是另有原因?
临床资料
患者男性,39岁,因“右上肢无力1年半,加重4个月”于2007年9月5日入院。患者1年半前无明显诱因逐渐出现右手握物费力,抬臂尚正常,未感麻木及其他肢体无力。
近4个月来上述症状加重,右手屈指、伸指均困难,腕、肘、肩屈伸均有力、关节活动不受限,伴鱼际肌、小鱼际肌及前臂肌轻度肌萎缩,不伴肌肉酸痛及肉跳感,余肢体活动好,病程中有间断轻度头痛,偶影响睡眠,但无恶心、呕吐,无肢体抽搐。自发病以来一般情况好,饮食二便正常,体重无减轻。既往:23年前外伤致“右顶叶硬膜外血肿”行清除术,术后良好。曾长期居住内蒙古自治区牧区,有牧犬密切接触史。
查体:一般情况好,右颅顶可见长7cm左右手术瘢痕,余内科检查无特殊所见。
神经科检查:神清语利,高级智能无减退,脑神经查体无异常,颈软无抵抗,右手鱼际肌、小鱼际肌、骨间肌和蚓状肌中度萎缩,右前臂肌群轻度萎缩,右上肢远端肌力减退2~4级(右拇指屈伸及内收、外展、对掌2级,四指屈3级,伸2级;前臂屈伸及内外旋4级,腕部屈伸及伸直后内收、外展均4级),近端肌力5级,右侧肱二、三头肌及桡骨骨膜反射(++++),右Hoffmann征(+),右Rossolimo征(+),右上肢自肩以下痛触觉较对侧明显减退。其他三肢的肌力、肌张力、腱反射、深浅感觉均正常,病理征未引出。
辅助检查:血尿便常规、肝肾功能、血糖、肌酶谱、ESR、肿瘤标志物均正常。肌电图(2007‐8‐21):①右外展拇短肌插入电位延长,右外展拇短肌、右外展小指肌、右第一背侧骨间肌出现纤颤电位(++)和正向电位(++);②双侧胸锁乳突肌、右肱二头肌、右伸指总肌、右屈指浅肌、左外展拇短肌、左第一背侧骨间肌肌电图均正常;③双侧正中神经、尺神经的运动神经和感觉神经传导速度、远端潜伏期皆正常范围(表2.5‐1)。
结论:“右上肢、右手诸肌神经源性损害;双侧正中神经和尺神经运动、感觉神经传导速度、远端潜伏期及波幅均正常”。颈椎MRI(2007‐8‐27):“C5~6椎间盘偏左后方轻度突出,硬膜囊轻度受压”。头颅CT(2007‐9‐5):“左额叶、额顶深部、左顶叶多发类圆形低密度,顶叶有一横行分隔,额叶病灶呈分叶状,其内可见点状高密度影,周围无明显水肿”。头颅MRI(2007‐9‐6)及增强(2007‐9‐10,图2.5‐1):“左侧大脑额叶、顶叶实质内多发囊实性类圆形长T1、长T2病灶,边界光滑清晰;病灶内信号欠均匀,有结节状稍低T1、等T2液化灶;增强后病灶中央呈不规则长T1信号强化;病灶内有点状钙化及小灶性坏死区,外周无明显水肿及占位征象,左侧脑室前脚相对稍窄,中线结构无移位”。
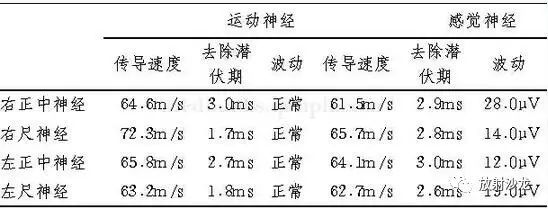
表2﹒5‐1双侧正中神经、尺神经的神经传导速度
初步诊断:
①手肌萎缩原因待查:顶叶性肌萎缩可能性大,肌萎缩侧索硬化、颈椎病待除外;
②颅内占位病变性质待查:脑包虫病可能性大,脑肿瘤待除外。进一步检查及治疗:2008年9月20日行开颅探察术,对三处肿瘤组织都进行了大部清除术。病理结果:肿瘤性星形细胞,体积小,胞突细长,含大量胶质纤维,基质可见黏液变性和微囊形成(图2.5‐2A、B);免疫组化GFAP(+++,图2.5‐2C)。诊断:弥漫型星形细胞瘤(WHOⅡ级),原浆型星形细胞瘤亚型。
讨论
本例患者中年男性,慢性进行性病程,临床表现仅为右上肢无力,手肌萎缩,体检发现右上肢呈上运动神经元病损,合并感觉障碍。首先,与肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)进行临床鉴别,ALS一般属于泛发性疾病,而该例仅为单侧肌萎缩,且有单侧感觉障碍,较难解释。ALS属全身上下运动神经元病变,呈慢性病程,不会始终局限于单侧上肢,且本例虽然肌电图出现右手肌异常电位,但无胸锁乳突肌等广泛运动神经元受累[1]。
客观查体存在右上肢单肢的感觉减退,这种感觉减退常是大脑皮质的损害特点。故可排除ALS诊断。另外,须注意与脊肌萎缩症(远端型)鉴别,结合影像学检查及存在右上肢感觉障碍,该病难以成立。其三,与颈椎病(神经根型或脊髓型)相鉴别,从影像学上看,颈椎间盘轻度突出偏于左侧,而该例为右侧上肢肌萎缩且呈上运动神经元损害。从感觉障碍分析,本例呈皮质型,而非神经根型障碍,故颈椎病性肌萎缩可以除外。
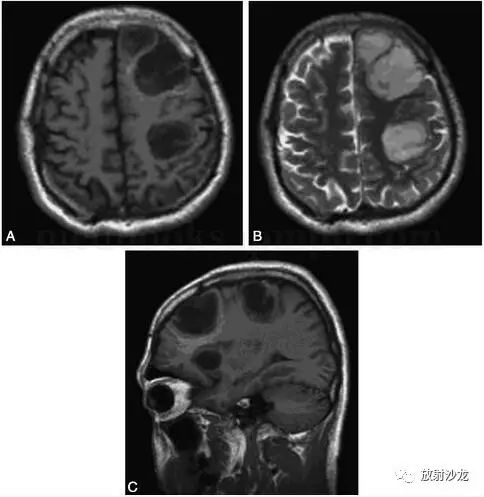
图2﹒5‐1MRI
左额叶、顶叶实质内三处类圆形长T1、长T2病灶;额叶病灶内信号不均,有结节状稍低T1、等T2液化灶A﹒T1WI;B﹒T2WI;C﹒矢状位T1WI

图2﹒5‐2肿瘤细胞体积小,基质变性
A﹒HE,100×;B﹒HE,200×;C﹒免疫组化GFAP(+++),400×
本例影像表现为脑多发囊样占位病灶,有典型原浆型星形细胞瘤影像表现:CT为均匀低密度灶,内有钙化及囊性变,囊内出血及小片状坏死改变;MRI病变呈T1WI低信号、T2WI高信号灶并有不同程度周边及囊内不规则强化,从CT或MRI影像特点方面及患者有牧区犬类接触史,还应与脑包虫病鉴别,但脑包虫病一般为单个囊样水样密度,多发灶极少,增强扫描不加强,同时血嗜酸性粒细胞明显增高等特点可以排除。
同心圆性硬化(Balo病)的首发症状常以精神智能或性格改变为主,定位体征少且不稳定,MRI在数月后仍出现“同心圆”或“煎蛋样”改变,边缘部分强化不影响囊内影像,对比本例特点可以排除本病。最后,颅内淋巴瘤、结核瘤、脑脓肿、某些肉芽肿及脑转移瘤[2],也多少有相似的影像学改变,在影像分析时应予以考虑。因此影像检查对本例的定位定性诊断有一定帮助。结合病理的回顾性分析本例幕上的星形细胞瘤或间变性星形细胞瘤的影像学表现还是比较典型的。
最近解剖生理学认为:并非所有来源于丘脑的感觉传入纤维都终止于感觉皮质,而有一部分终止于中央前回的运动皮质内,感觉性和运动性皮质区有部分重叠,总称之为“感觉运动区”。此区的某些感觉性冲动可以转换成运动性冲动,谓之“感觉运动反馈环路”。在此环路内,其锥体束纤维大多不需要中间神经元而直接终止于脊髓的前角细胞,影响前角功能。因本例系顶叶性肌萎缩且伴有上下运动神经元性的右上肢远端肌萎缩[3],用此理论可以直接解释其肌电图有少量失神经电位,而周围神经传导速度正常的表现。
本病例手部肌肉萎缩的原因是肿瘤组织压迫顶叶皮质运动区所致的“顶叶性肌萎缩”。顶叶性肌萎缩病因多见于外伤、脑血管病、多发性硬化等病,多为肢体远端肌受累。肿瘤引起者少见,检索Medline,国内未见报道,国外报道至今查到6例[4~7]。原浆型星形细胞瘤发病率并不低,占所有星形细胞瘤的10%~15%。多位于额颞叶、脑干、小脑。但未检索到原浆型星形细胞瘤引起顶叶性肌萎缩的国内外报道。
结合本病例总结肿瘤引起顶叶性肌萎缩的特点:
①引起肌萎缩病程约1~3年,多为良性胶质细胞瘤。考虑因为原浆型星形细胞瘤等良性肿瘤分化高、生长缓慢,在引起颅内压增高、癫痫等脑部症状前,先出现运动区相应肌肉的失用性萎缩;
②多为肢体单瘫,上肢[4~6]多于下肢[7],与上肢在顶叶皮质占有更大面积更易受累有关;
③肌电图无[4]或仅有少量失神经电位[5],而周围神经传导速度均正常[5];
④肌活检正常或发现明显选择性Ⅱ型肌纤维的萎缩[5]。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









