
Nat Commun:王建新教授团队建立基于簇分解和异常检测算法的罕见细胞识别方法
2024-09-04 测序中国 测序中国 发表于上海

基于单细胞转录组测序数据,提出了准确的罕见细胞识别算法,并开发了相应的软件scCAD。
单细胞转录组测序(scRNA-seq)技术已经成为描绘复杂组织中细胞景观的关键工具。技术革新所带来的数据规模的增长有助于识别和分析在特定生物学或病理状态下扮演着重要角色的罕见细胞类型。准确识别这些罕见细胞对于全面理解组织功能和结构、揭示新的生物学现象或疾病机制至关重要。然而,识别这些罕见细胞的任务面临显著挑战,主要体现在以下两个方面:(1)罕见细胞在总体细胞群体中的比例极小,其基因表达差异性往往不足,导致其在整体细胞群体中难以被准确识别;(2)单细胞转录组测序数据本身具有较高的噪声和稀疏性,这可能掩盖罕见细胞的特征,从而进一步影响其准确识别。近年来,尽管提出了许多用于识别罕见细胞的方法,这些方法往往依赖于基于部分或整体基因表达的单次聚类结果,这可能导致罕见细胞被忽视,从而限制了其准确识别。此外,这些方法通常缺乏在广泛研究的不同组织和物种中的单细胞数据上的深入验证,未能充分证明其准确性和鲁棒性。为了更好地识别和理解罕见细胞,迫切需要发展更加精准和全面的识别方法。
2024年8月31日,中南大学计算机学院王建新教授团队在Nature Communications上在线发表题为“scCAD: Cluster decomposition-based anomaly detection for rare cell identification in single-cell expression data”的研究论文,基于单细胞转录组测序数据,提出了准确的罕见细胞识别算法,并开发了相应的软件scCAD。中南大学计算机学院徐云佩、滑铁卢大学计算机系王劭恺为论文共同第一作者,中南大学计算机学院李洪东副教授为共同通讯作者,中南大学为第一署名单位,该研究受国家重点研发计划、国家自然科学基金、湘江实验室揭榜挂帅项目等多个项目支持。

文章发表在Nature Communications
scCAD首先采用了一种集成特征选择方法,以有效地保留罕见细胞类型的特异性信号。不同于传统方法仅依赖于高变基因(HVGs)进行分析,scCAD还通过利用基于全局基因表达的细胞预聚类标签和随机森林模型,识别出高重要性的基因(HIGs)。接下来,scCAD通过对每个簇中最具差异性的信号进行迭代聚类,分解初始聚类中的主要簇。在簇分解之后,罕见细胞类型主导的簇数量显著低于主要细胞类型主导的簇数量。为了提高计算效率,scCAD通过合并部分相邻簇来减少所分析簇的数量。最后,scCAD基于每个簇的差异表达基因(DEGs)和孤立森林模型计算独立性评分,以衡量簇的罕见性。图1展示了scCAD的示意流程图。
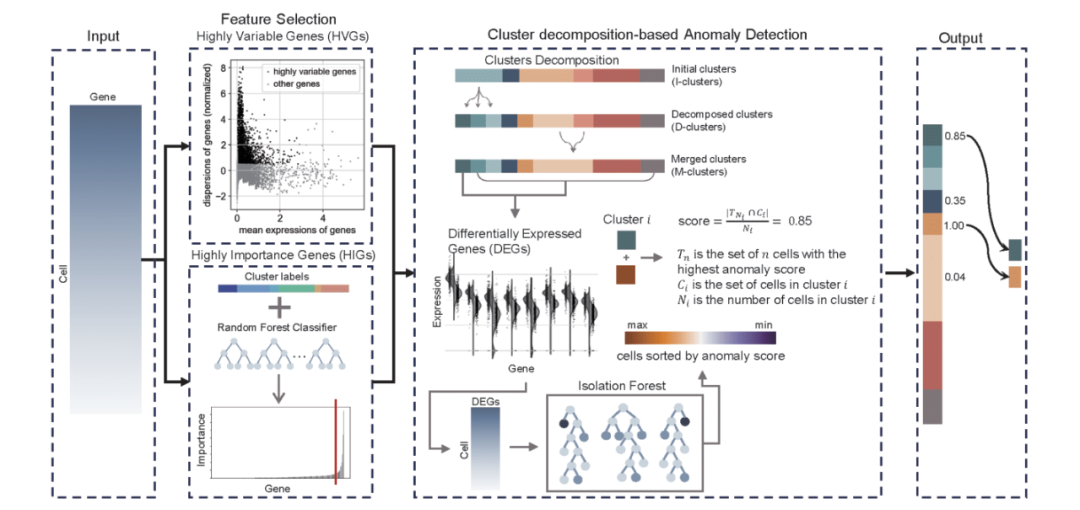
图1. scCAD流程图
研究团队将scCAD与十种最先进的方法进行了比较,以评估其在识别代表多种生物学场景的25个真实单细胞RNA测序数据中的罕见细胞类型方面的表现。评估使用了罕见细胞类型的F1分数。如图2a所示,scCAD在整体性能上表现最佳,F1分数达到0.4172,较第二名和第三名的方法(SCA: 0.3359, CellSIUS: 0.2812)分别提高了24%和48%。为了进一步评估这些算法的效果,研究团队计算了每种方法成功识别至少一种罕见细胞类型的数据集总数。如图2b所示,scCAD在20个数据集中成功识别了罕见细胞类型。
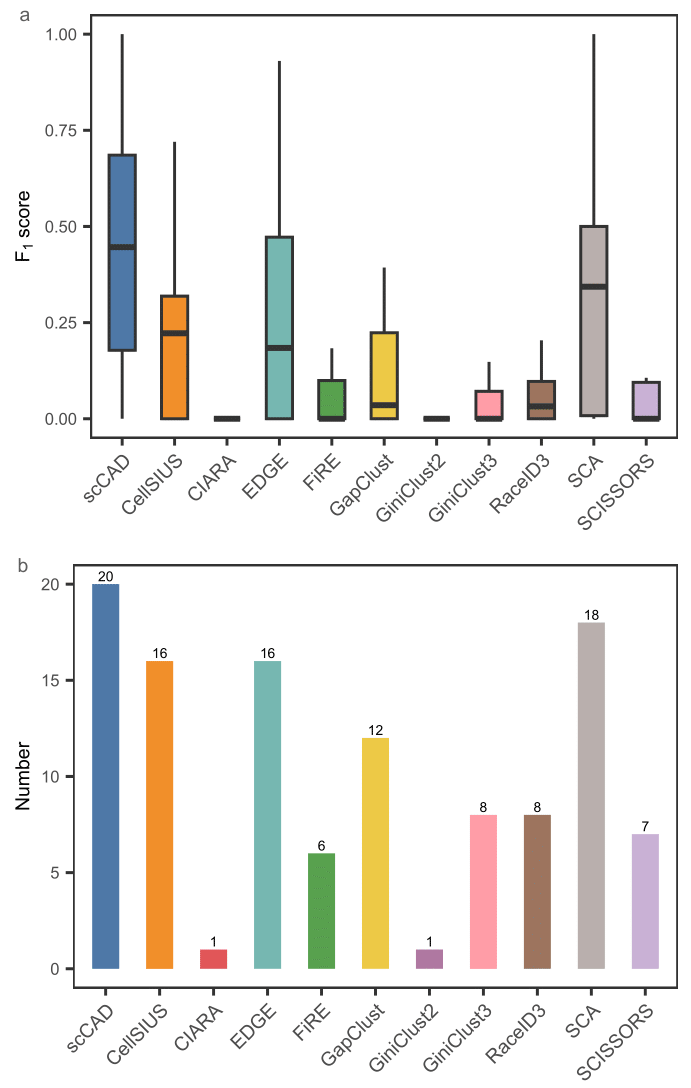
图2. scCAD与10种最先进方法在25个真实数据集上的基准测试对比结果
肺部气道是哮喘等疾病的重要部位,其中罕见细胞在维持气道功能方面起着关键作用。研究团队在小鼠气道上皮的单细胞转录组测序数据中应用了scCAD。通过该方法,共识别出了三个罕见细胞簇,分别为R1(0.42%)、R2(0.26%)和R3(0.57%)(见图3b)。为了验证这些簇的真实性,团队首先从数据的原始研究中获取了罕见细胞类型的注释信息,然后将这些已注释的罕见细胞类型的差异上调基因的表达与scCAD鉴定的细胞簇中的基因表达进行比较,如图3c所示。结果显示,簇R2和R3分别对应离子细胞和杯状细胞,这两种细胞类型在数据集中分别仅占0.90%和0.36%。具体来说,R2簇中的细胞表现出经典的离子细胞标志物,例如转基因Foxi1-EGFP、V-ATP酶亚基基因Atp6v0d2、囊性纤维化跨膜传导调节因子(Cftr)基因、转录因子Ascl3以及Smbd1(又名Gm933)。R3簇中的细胞则展现了与“Goblet-1”杯状细胞亚型相关的经典标记物,特别是富含编码关键粘膜蛋白(如Tff1)和分泌调节因子(如Lman1l)的基因表达。其他方法的可视化结果表明,只有scCAD能够同时准确识别离子细胞和杯状细胞。相比之下,簇R1则由30个被注释为Club类型的细胞组成。对簇R1及其他细胞类型中R1簇特异性上调基因的表达进行了可视化(图3d),结果显示这些基因在其他细胞类型中并未显著表达。值得注意的是,R1簇与原始文献作者Montoro等人在分析细胞分化轨迹时发现的“小丘”细胞具有惊人的相似之处,这些罕见的过渡细胞通过Krt13和Krt4的独特表达将基底细胞与Club细胞连接起来。
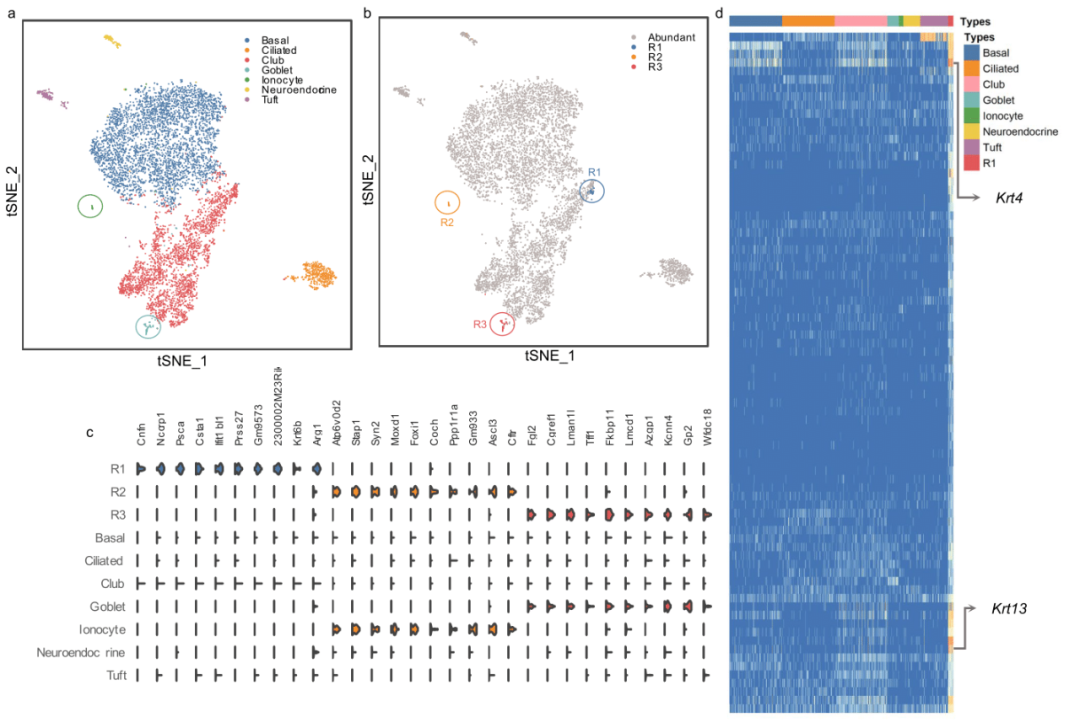
图3. scCAD在小鼠气道上皮数据中的识别结果的可视化分析
树突状细胞(DC)在病原体感应、吞噬作用和抗原呈递中扮演着核心角色。然而,树突状细胞是最罕见的免疫细胞类型之一,仅占外周血单核细胞(PBMC)的1-2%。Villani等人通过荧光激活细胞分选(FACS)分析了树突状细胞的表达谱,识别出了六种不同的树突状细胞亚型。为了进一步验证scCAD识别的罕见簇的可靠性,研究团队将scCAD应用于广泛使用的68k PBMC数据集,探讨是否能够识别原始注释中未捕获的树突状细胞亚型。如图4b所示,scCAD识别出了四个罕见细胞簇,分别为R1(0.50%)、R2(0.24%)、R3(0.13%)和R4(0.12%)。簇R2主要由巨核细胞组成,这种细胞类型在数据集中仅占0.4%。此外,R1和R4主要由原始研究中注释的树突状细胞组成,而R3则主要由CD19+ B细胞构成。为了探究R1和R4这两个树突状细胞簇的真实身份,研究团队计算了分别在两个不同的数据中,这些簇与经过充分验证的树突状细胞亚型在同一标记基因集上的平均表达之间的相关性。如图4c所示,R1和R4与树突状细胞亚型DC1和DC6(pDC)的相似性最高,分别为0.8和0.74,显著高于其他亚型。图4d展示了簇R1、R4以及所有注释为树突状细胞类型的细胞在不同树突状细胞亚型的标记基因表达分布上的情况。属于亚型DC1和DC6的标记基因在簇R1和R4中均表现出了显著表达。综合图4c和4d的结果,可以确定簇R1中的细胞属于亚型CLEC9A+ DC,而簇R4对应于pDC。尽管簇R3中的细胞最初被注释为CD19+ B细胞,研究团队还是鉴定出了多个浆细胞的标志物,如CD27(TNFRSF17)、MZB1、DERL3、ITM2C和IGLL5。其他研究也报告了该数据集中浆细胞的存在且高度罕见,进一步验证了研究团队的发现。
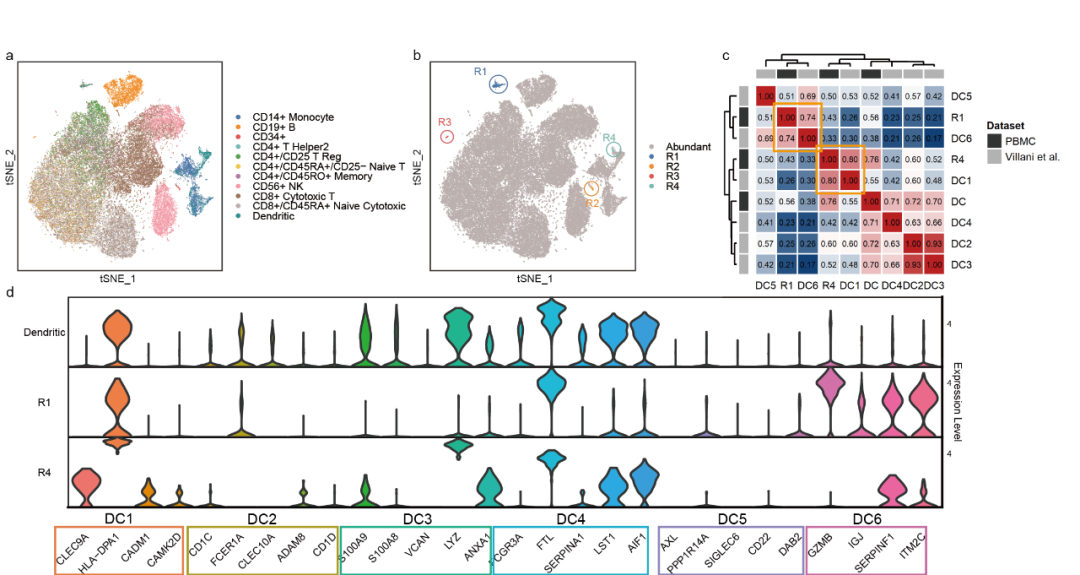
图4. scCAD在68k PBMC数据的识别结果的可视化分析
此外,研究团队还对其他单细胞转录组测序数据进行了深入分析,特别是在透明细胞肾细胞癌的数据中,scCAD有效纠正了罕见细胞的注释错误,并识别了与疾病相关的免疫细胞亚型。Kaplan-Meier分析表明,所鉴定的罕见细胞簇中的差异表达基因与总生存期显著相关,提供了有价值的预后信息。此外,对两个大规模免疫学数据集的分析进一步证明了scCAD的良好可扩展性。
综上所述,研究团队提出了一种基于簇分解和异常检测的罕见细胞识别方法。该方法在25个真实的单细胞转录组测序数据集上表现卓越,优于10种先进的现有方法。通过涵盖小鼠气道、大脑、肠道、人类胰腺、免疫学数据以及透明细胞肾细胞癌等多个生物场景的案例研究,scCAD成功识别了以往研究中报告的罕见细胞类型,验证了其稳健性和鲁棒性。
文章第一作者:
徐云佩,中南大学计算机学院博士。主要研究领域为生物信息学,包括单细胞多组学数据分析,开发了scCAD、CellBRF等单细胞转录组测序数据分析工具,在Nature Communications,Bioinformatics,Briefings in Bioinformatics,IEEE/ACM TCBB等期刊,ISMB、BIBM等生物信息学会议发表多篇文章。
王邵恺,博士毕业于滑铁卢大学计算机系,现为香港科技大学博士后。主要研究领域为生物信息学,包括基于质谱的蛋白测序技术,肽的性质预测,单细胞与空间转录组学以及三代测序技术等,在Nature Communications, Briefings in Bioinformatics, IEEE JBHI, IEEE/ACM TCBB, Analytical Chemistry等期刊发表多篇文章。
参考文献:
Xu, Y., Wang, S., Feng, Q. et al. scCAD: Cluster decomposition-based anomaly detection for rare cell identification in single-cell expression data. Nat Commun 15, 7561 (2024). https://doi.org/10.1038/s41467-024-51891-9
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#单细胞转录组测序# #罕见细胞#
66