Genome Biology:癌症突变组学特征数据冗余普遍存在!简单的多组学集成无法有效提高突变预测性能
2022-07-30 测序中国 测序中国

多组学建模分析结果表明,由于基因表达和DNA甲基化捕获的突变状态信息是高度冗余的,添加数据类型并不会导致分类器性能的提高。
癌症可以由多种不同的基因突变引发、驱动,但这些突变往往集中于有限的通路或信号传递过程。由于驱动基因突变提供的预后信息有限,全面了解不同的基因突变如何干扰中枢通路对精准医疗和识别特异性生物标志物至关重要。 癌症基因组图谱(The Cancer Genome Atlas, TCGA)项目中的泛癌图谱为33种癌症类型的数万个样本提供了统一处理的多平台组学数据。在这些公开数据的支持下,越来越多的研究将癌症中驱动基因突变与下游基因表达变化关联,用于探究遗传变异的功能效应。虽然蛋白质组学数据可以更直接地对应某些癌症表型和通路异常,但肿瘤细胞系中基因表达和蛋白质丰度之间的相关性有限。因此,整合不同的数据模式或结合多种数据模式可能比仅仅依靠基因表达作为功能特征鉴别依据更有效。但基于目前的突变相关数据集,真的是这样吗?
近日,美国科罗拉多大学医学院的研究团队在Genome Biology发表了题为“Widespread redundancy in -omics profiles of cancer mutation states”的文章。研究团队比较分析了TCGA泛癌症图谱中的组学数据类型,并评估其作为癌症基因突变的多变量功能读数(readouts)的作用。分析结果表明,相对于癌症类型校正基线,基因表达数据能够对大多数基因的突变状态提供良好的预测;对于多数基因而言,多种数据类型几乎具有同等有效的预测能力。与使用单一数据类型的性能最高的模型相比,将数据类型组合到单个多组学模型中进行突变预测的方法几乎没有性能优势。这一研究结果对未来指导癌症功能基因组学的研究具有深远意义。

主要研究内容
研究团队从TCGA泛癌图谱的癌症样本中收集了五种不同的数据类型,包括基因表达数据(RNA-seq)、DNA甲基化(27K和450K)、蛋白质丰度(RPPA数据)、microRNA表达数据和体细胞突变数据。为了将这些不同的数据类型与突变状态的变化相关联,研究团队使用弹性网络回归算法预测癌症基因中是否存在突变,并将readouts作为预测特征(图1)。在泛癌环境中,研究团队评估了所得的突变状态分类器,并比较了不同数据类型的预测性能。
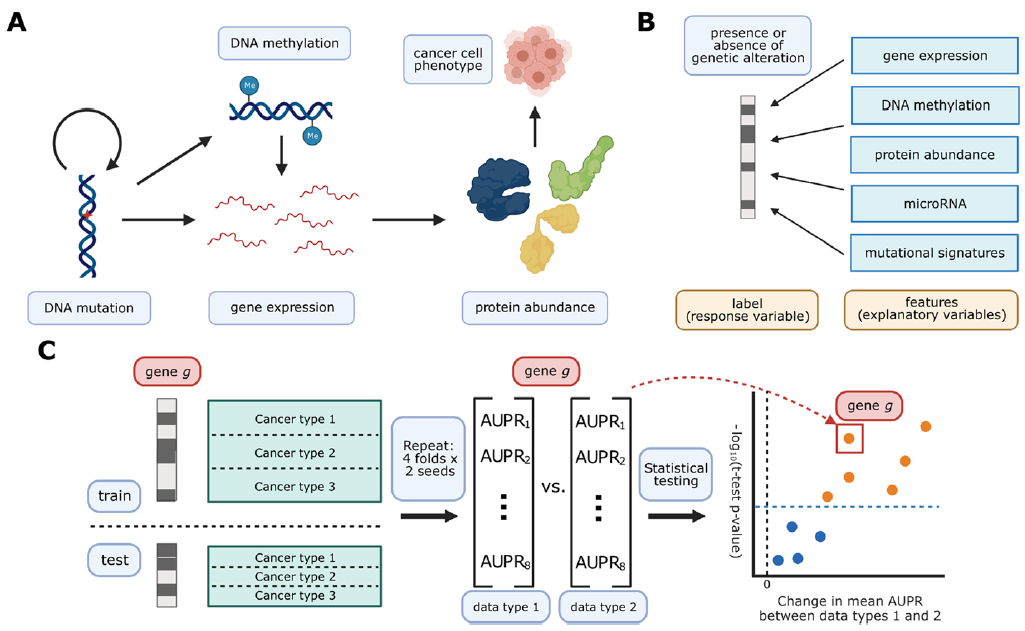
图1. 整体研究模式图。来源:Genome Biology
研究团队评估了几种不同基因集的基因表达数据对突变状态的预测性能,并将其作为基线。先前实验已评估了TCGA中前50个最容易突变的基因,此次,研究团队试图将其扩展到更广泛的基因集列表中(图2)。为评估使用已知的癌症相关基因是否有助于提高预测性能,研究团队从前期的研究结果和数据库中总结了268个癌症相关基因。
结果显示,来自癌症相关基因集的基因比随机选择的基因或通过总突变数选择的基因更具可预测性。选定的癌症相关基因集中约45%的基因具有统计学显著的可预测性;随机基因集中仅有5.22%的基因、突变最多的基因集中29.9%的基因有显著可预测性。上述结果表明,依据对目标基因参与的癌症途径和过程的先验知识来选择突变预测的目标基因,而不是通过随机或仅基于突变频率,可以提高预测性能,能够从基因表达数据中识别出具有更高可预测性的突变。
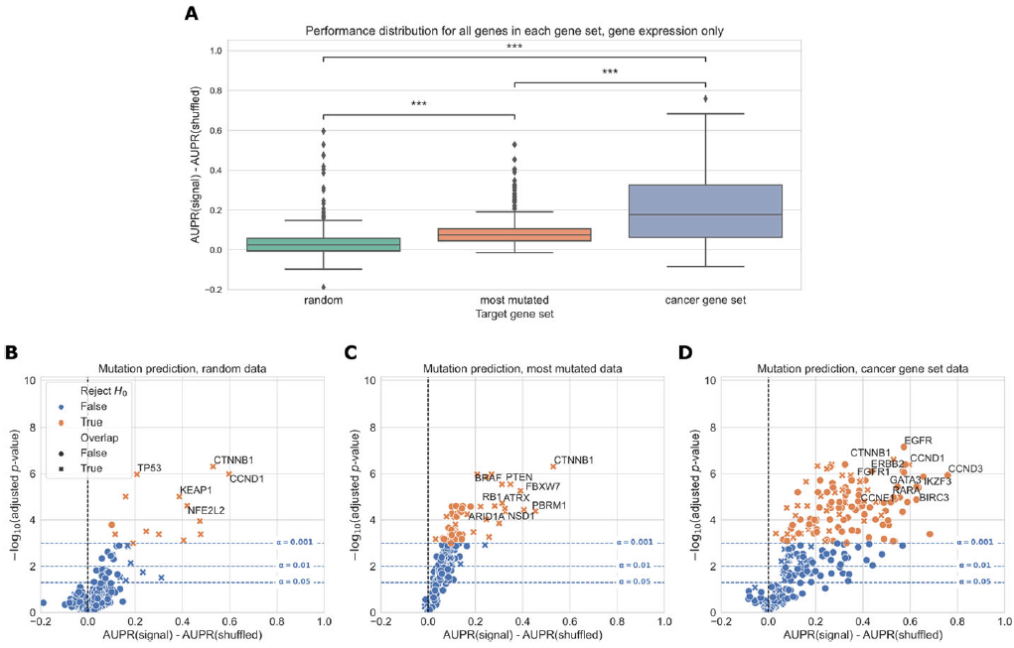
图2.三个基因集的总体性能分布。来源:Genome Biology
比较不同数据集的预测性能
接下来,研究团队比较了TCGA泛癌图谱中五种可用的功能数据类型(因为有两个DNA甲基化平台,所以共六个readouts)。在总结癌症相关基因集中的所有基因时观察到,与其他数据类型相比,基因表达数据往往能产生更好的预测。此外,在个体基因水平上,相对于置换基线,33/217个基因的突变可从RPPA数据显著预测,microRNA数据中有25/217个基因,突变特征数据中有2/217个基因。
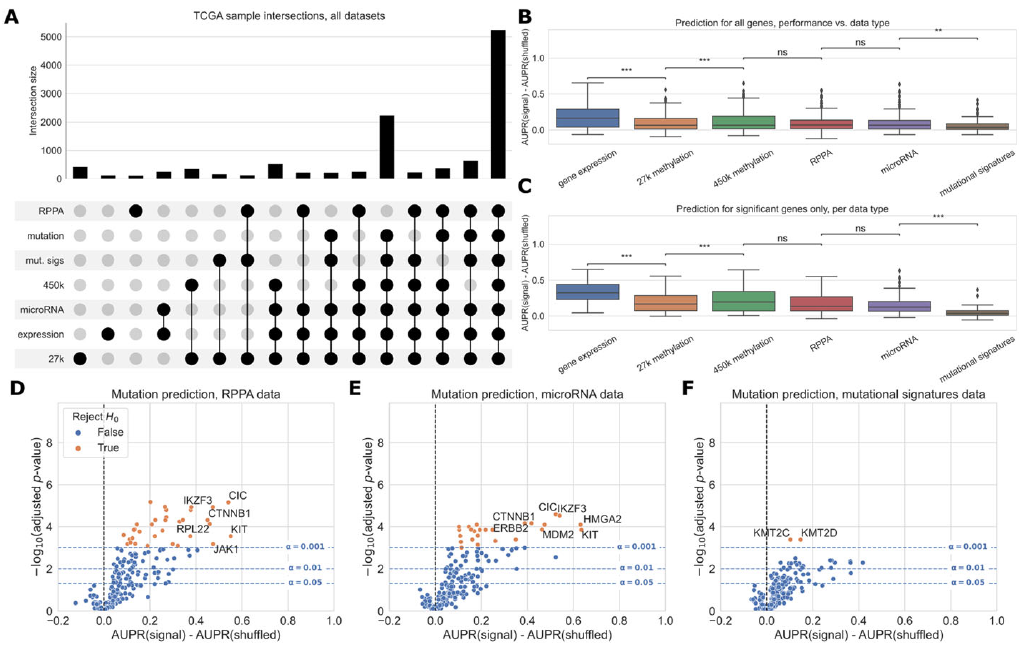
图3. 不同数据集的预测性能比较。来源:Genome Biology
在构建描述每个基因在不同数据类型中的预测性能热图时,研究团队发现许多基因可以被多种数据类型很好地预测。在至少被一种数据类型进行良好预测的86个基因中,60.5%能够被多种数据类型成功预测,这意味着多组学readouts包含相应基因中存在/不存在突变的可检测特征。上述结果表明,对于许多具有明确功能特征的强大驱动基因,不同的组学数据可以提供相似的信息内容,因此数据类型选择并不重要。在大多数情况下,相较不同的数据类型,这些基因往往是影响较大的预测因素。
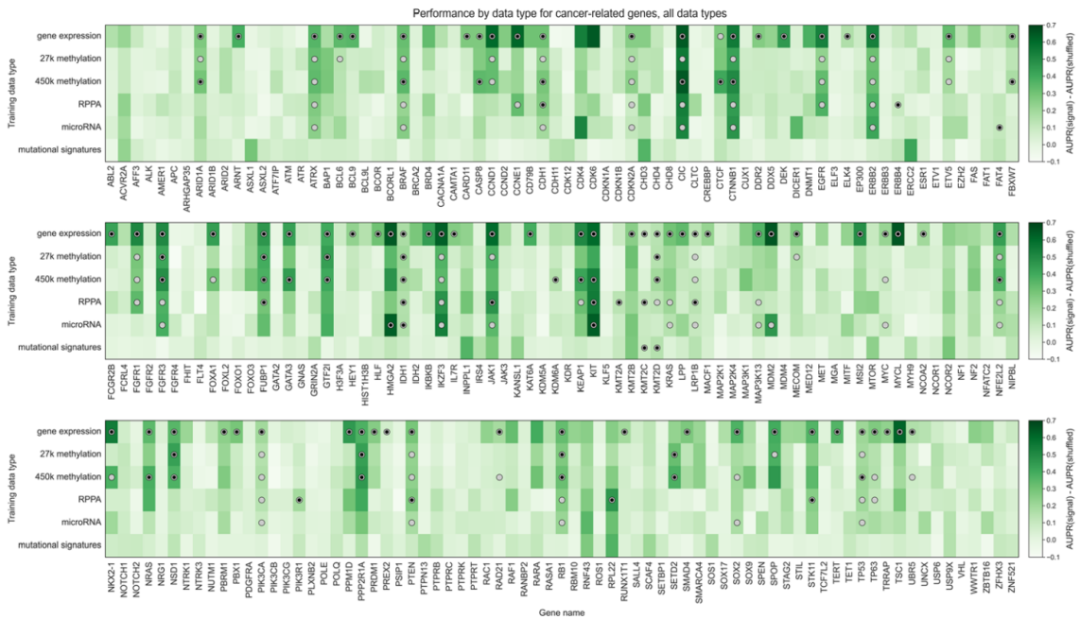
图4. 不同数据集的预测性能比较。来源:Genome Biology
最后,研究团队训练了“多组学”分类器,并在不同癌症类型中预测了6个经充分研究和广泛突变的驱动基因,即EGFR、IDH1、KRAS、PIK3CA、SETD2和TP53。对于6个目标基因,最好的单组分类器和最好的多组学分类器之间的性能相当,并无显著差异。此外,在不同的分类器和数据类型中,研究团队发现了基于目标基因的不同模式。例如,对于IDH1和TP53,无论数据类型如何,性能都是相对一致的,这表明其基线性能较高,数据的增加几乎没有改进的空间;对于EGFR、KRAS和PIK3CA,整合基因表达和甲基化数据的预测性能与基因表达数据的预测性能相同或更差。总体而言,与最佳单个数据类型相比,以相对简单的方式组合数据类型,几乎没有改善预测能力。
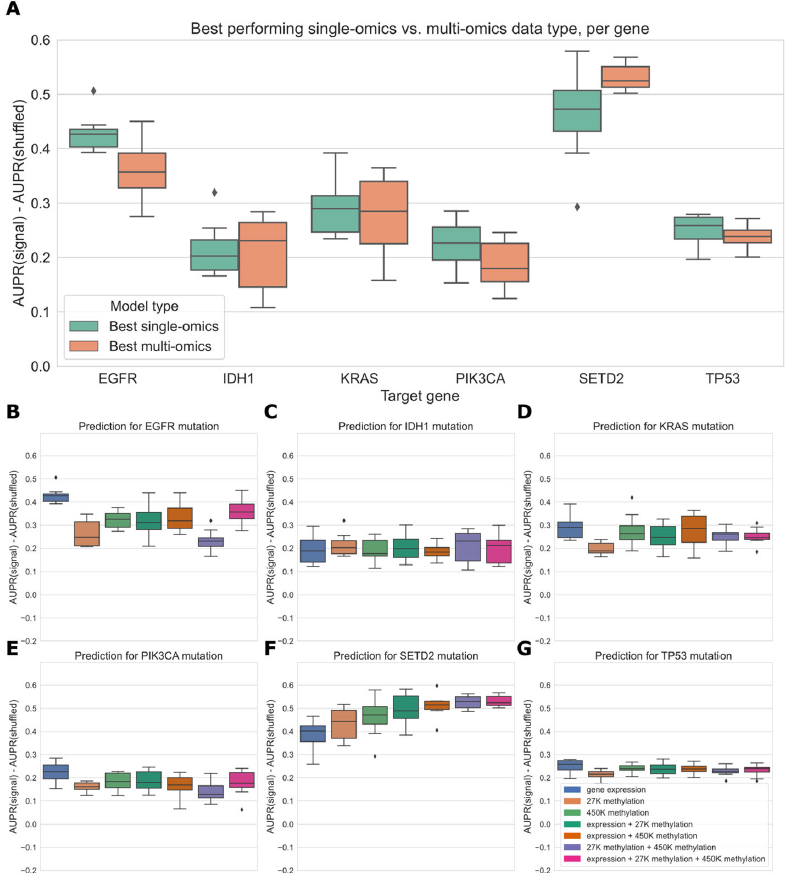
图5. 单数据和多组学数据预测性能的比较。来源:Genome Biology
结 语
综上所述,该研究对TCGA泛癌症图谱中的数据类型进行了大规模比较,并整合了不同癌症类型和驱动基因的结果。结果显示,相对于基线模型,基因表达数据可以最有效地捕捉突变状态的特征。此外,多组学建模分析结果表明,由于基因表达和DNA甲基化捕获的突变状态信息是高度冗余的,添加数据类型并不会导致分类器性能的提高。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#预测性能#
58
#Biol#
119
#Bio#
52
学习#肿瘤#
52