
Nat Commun:王建新/罗峰/肖传乐建立基于第三代测序数据的二倍体组装算法
2024-04-10 测序中国 测序中国 发表于陕西省

基于第三代测序数据,提出了二倍体的单倍型组装新算法,并开发了相应的软件PECAT。
基因组组装是基因组学研究的一项基本任务。针对第三代测序技术的长读数(牛津纳米孔测序和PacBio单分子实时测序),目前已有许多组装工具能够使用它们有效地重建单倍体或近交物种的高质量基因组。然而,这些工具无法有效组装二倍体的基因组,生成单倍型混合的组装结果,存在许多单倍型转换错误,并且丢失很大一部分遗传信息。由于长读数(如PacBio CLR读数和Nanopore读数)通常包含5-15%的测序错误,因此组装算法很难将单倍型差异信息与测序错误区分开来,无法生成更长的单倍型组装结果。
结合父母本读数、HiFi读数、Hi-C读数、Strand-seq测序数据或者配子测序数据的多数据联合组装,能够实现更连续的单倍型组装结果,但其要求额外的测序数据,提高了测序成本也限制了这些方法的应用。高精度的PacBio HiFi读数(<1%的错误率)更容易识别单倍型差异,已被广泛用于单倍型组装中。但HiFi读数的平均长度(10-25kb)短于错误率较高的长读数,如纳米孔的超长读数长度高达1M,读数N50>100kb。较长的读数通常有助于获得更连续的组装结果,目前的组装算法未能充分利用这些优势实现更连续的单倍型组装结果。
2024年4月5日,中南大学计算机学院王建新教授、美国克莱姆森大学计算机学院罗峰教授和中山大学中山眼科中心肖传乐副研究员在Nature Communications上在线发表题为“De novo diploid genome assembly using long noisy reads”的研究论文,基于第三代测序数据,提出了二倍体的单倍型组装新算法,并开发了相应的软件PECAT。中南大学计算机学院聂藩、倪鹏为论文共同第一作者,中南大学为第一署名单位,该研究受国家重点研发计划、国家自然科学基金、湘江实验室揭榜挂帅项目等多个项目支持。

文章发表在Nature Communications
PECAT采用先纠错再组装的策略,分为纠错步骤和组装步骤,而组装步骤采用两轮组装策略,如图1所示。在纠错步骤中,PECAT识别测序错误和单倍型差异在纠错图(POA图)上的区别,选择性与被纠错读数来自相同单倍型的读数参与纠错,从而保证纠错读数单倍型信息的一致性和完整性(图1a)。完成纠错后,PECAT执行第一轮组装,生成单倍型混合的组装结果,如图1b所示。在第二轮组装中,将读数比对到混合组装结果上,识别读数携带的SNP信息,通过局部聚类方式识别单倍型不一致的重叠关系,过滤不一致的重叠关系后再次组装,生成单倍型组装结果(图1c)。
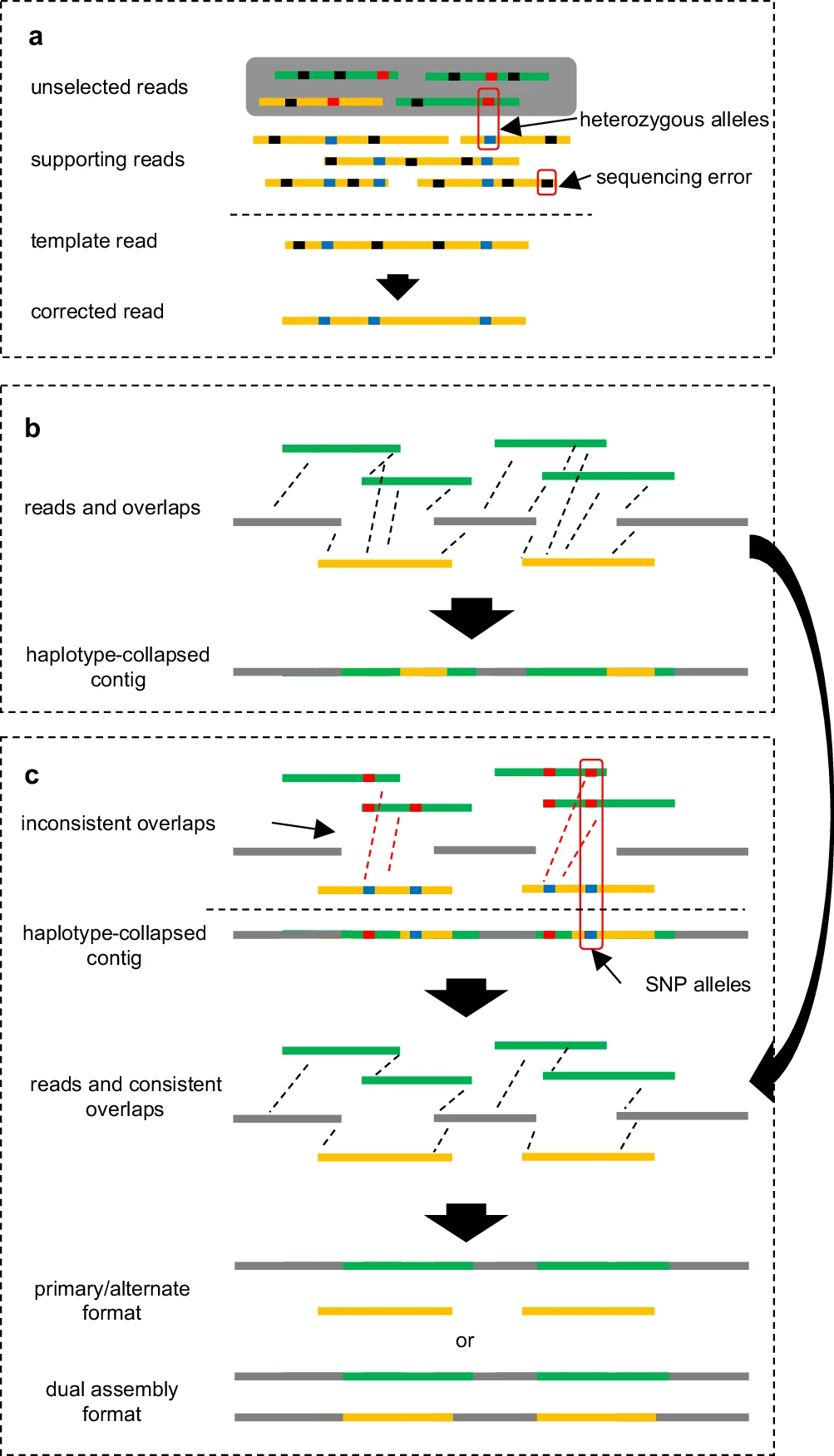
图1. PECAT组装算法框架图
研究团队首先对PECAT的纠错能力进行了测试,评估其纠正测序错误和保留单倍型差异信息的能力。如图2a所示,在模拟的二倍体数据上,不同纠错算法的准确率类似,均超过99%,但PECAT算法的SNP位点的准确率在不同杂合度下都超过91%,而其它算法都小于81%。图2b显示,在HG002的difficult-to-map和low-complexity区域,PECAT纠错读数的准确率比NECAT的纠错结果高0.75%。在七个真实数据上(四个PacBio CLR数据和三个纳米孔数据上),PECAT纠错读数的单倍型信息的一致性和完整性超过其它纠错算法(图2c、d),特别是PECAT的单倍型信息一致性指标大于99.4%,而其它算法都小于94.8%。该结论也反映在图2e中,PECAT纠错读数的坐标更靠近坐标轴,说明其有效避免了单倍型信息的混合。
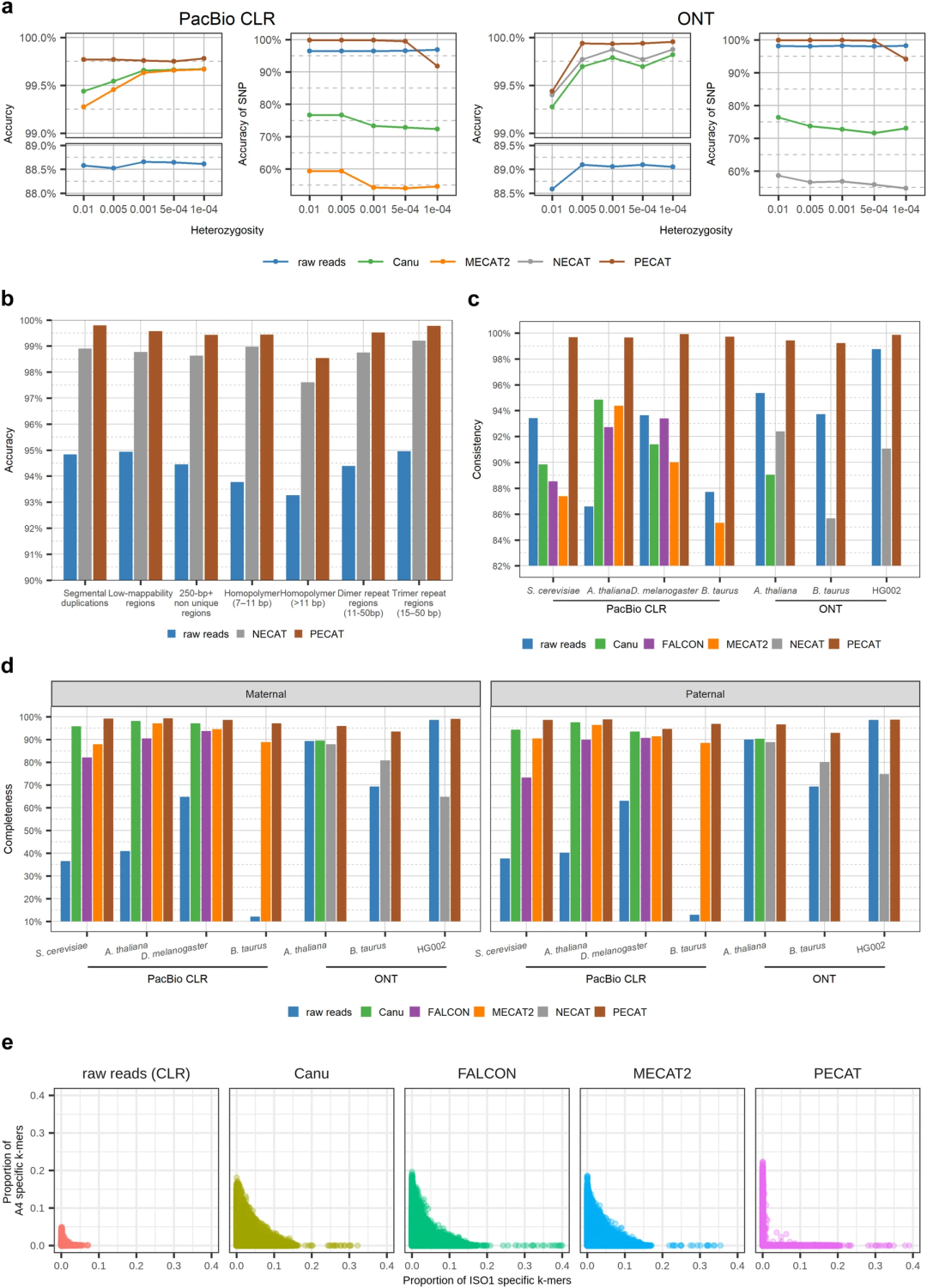
图2. 纠错性能评估
研究团队接着对PECAT的组装性能进行了评估。在七个真实数据上(四个PacBio CLR数据和三个纳米孔数据上),PECAT组装结果的NG50以及phase block NG50均超过其它工具。特别是在纳米孔的公牛数据(杂合度为1.48%)上,PECAT获得了单倍型几乎解决的组装结果,如图3a所示。团队评估了HG002组装结果的SNP、INDEL和SV的准确率和召回率,各个算法在纳米孔读数上的性能相近。在SNP和SV的结果上,纳米孔数据的组装结果与HiFi数据组装结果相近,但在INDEL的结果上,纳米孔数据的组装结果要差于HiFi数据的组装结果(图3b)。PECAT算法的特点能够有效避免第一轮组装的影响(大型的INDEL无法在第一轮组装中保留)。相比于基于抛光的二倍体组装算法,PECAT能够组装出二倍体之间大型INDEL(如图4所示),从而在基因组完整度(genome fraction)指标上超过其它工具,如图3c所示。
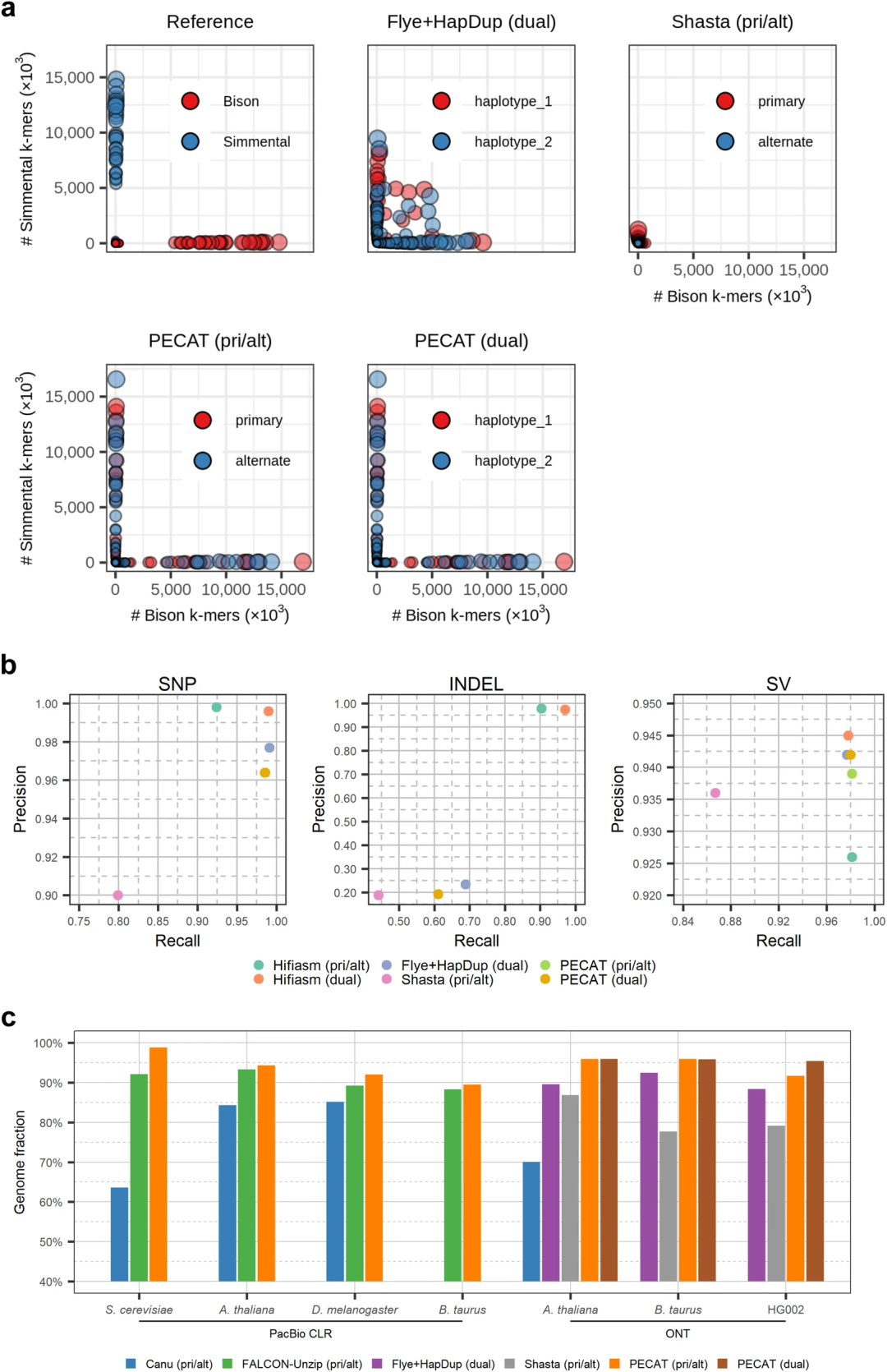
图3. 组装性能评估
随着测序技术的发展,测序数据的准确率进一步提高。研究团队测试了PECAT在准确率更高数据上的性能,如HiFi数据、纳米孔R10 数据和纳米孔R10 duplex数据。实验结果表明,PECAT同样能够有效地组装这些类型的二倍体数据。在HG002纳米孔R10 超长数据上,PECAT实现了phase block NG50为59.4/58.0 Mb的组装结果。上述结果表明,PECAT是一款高效、基于第三代长读数的二倍体组装工具。
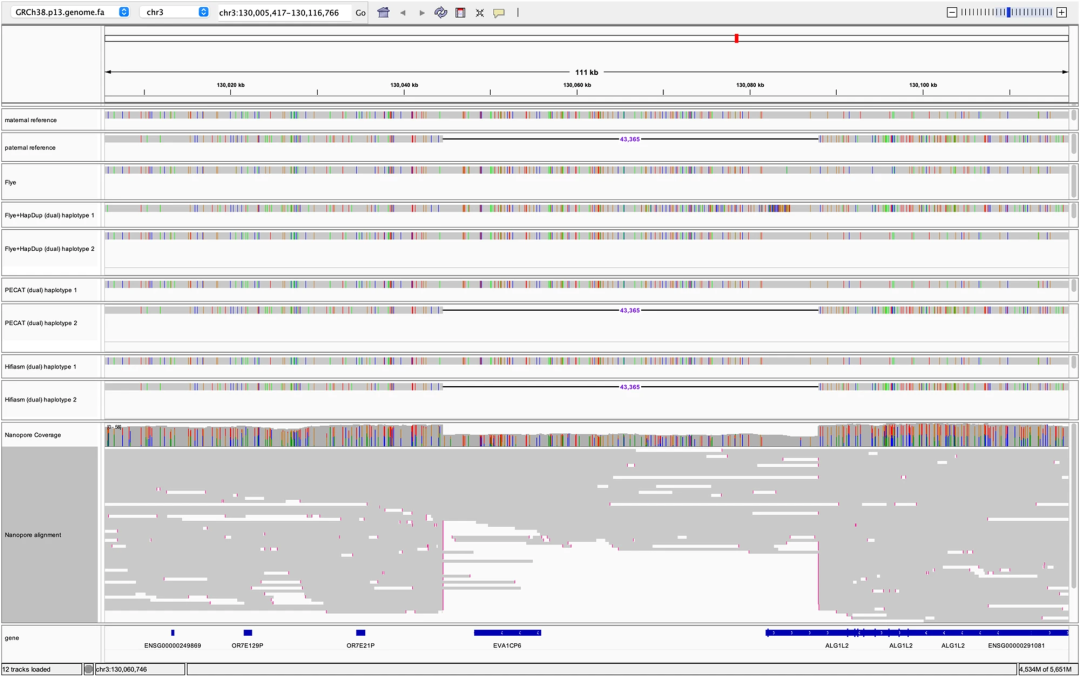
图4. HG002的大型INDEL的组装结果
综上所述,研究团队提出了基于第三代测序技术的二倍体组装算法PECAT。在不同数据集上的测试结果表明,PECAT能够克服测序错误的影响,有效区分二倍数据的单倍型,充分利用读数的长读数优势,在仅使用第三代长读数的条件下实现更连续的单倍型组装结果,为基因组学的多态性研究、物种进化研究、 疾病相关性研究等多种下游研究提供重要的支持。
文章第一作者:
聂藩,博士毕业于中南大学计算机学院,现为湘潭大学数学与计算科学学院讲师。主要从事三代测序基因组组装和分析研究,开发了NECAT、PECAT等三代测序基因组组装工具,在Nature Communications、Bioinformatics等期刊发表多篇文章。
倪鹏,中南大学计算机学院讲师。主要从事三代测序数据分析和疾病关联模式挖掘等研究,开发了甲基化检测工具DeepSignal、DeepSignal-plant、ccsmeth等,在Nature Communications、Bioinformatics等期刊发表文章10余篇。
参考文献:
Nie, F., Ni, P., Huang, N.et al. De novo diploid genome assembly using long noisy reads. Nat Commun 15, 2964 (2024). https://doi.org/10.1038/s41467-024-47349-7
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言












#测序# #二倍体组装算法#
65