ReactomeGSA:高效的单细胞通路分析工具
2021-02-06 周运来 简书

Pathway analyses是组学实验分析的关键方法。然而,整合来自不同组学技术和不同物种的数据仍然需要大量的生物信息学知识。在单细胞数据科学中,Pathway analyses也是基因表达数据链
Pathway analyses是组学实验分析的关键方法。然而,整合来自不同组学技术和不同物种的数据仍然需要大量的生物信息学知识。在单细胞数据科学中,Pathway analyses也是基因表达数据链接其他组学的主要路径,如单细胞转录组与代谢组学之间的链接。本文介绍的ReactomeGSA可以使用reacome数据库分析scRNA-seq数据。将来自来自不同物种的数据自动映射到一个共同的路径空间(基于生命科学底层的保守型)。将来自ExpressionAtlas和SingleCellExpressionAtlas的公开数据可以直接整合到分析中。ReactomeGSA大大降低了多组学、跨物种、比较通路分析的技术障碍。
通过ReactomeGSA R包的“analyze_sc_clusters”功能支持对scRNA-seq数据的分析,也可以通过直接从单细胞表达图谱导入。在这两种情况下,我们计算基因在一个簇内的平均表达量。这可以通过“Seurat”的AverageExpression函数,或者通过scater的“aggregateaccroscells”函数来完成,具体取决于输入对象。其实就是把数据库与富集方法封装到一起了。
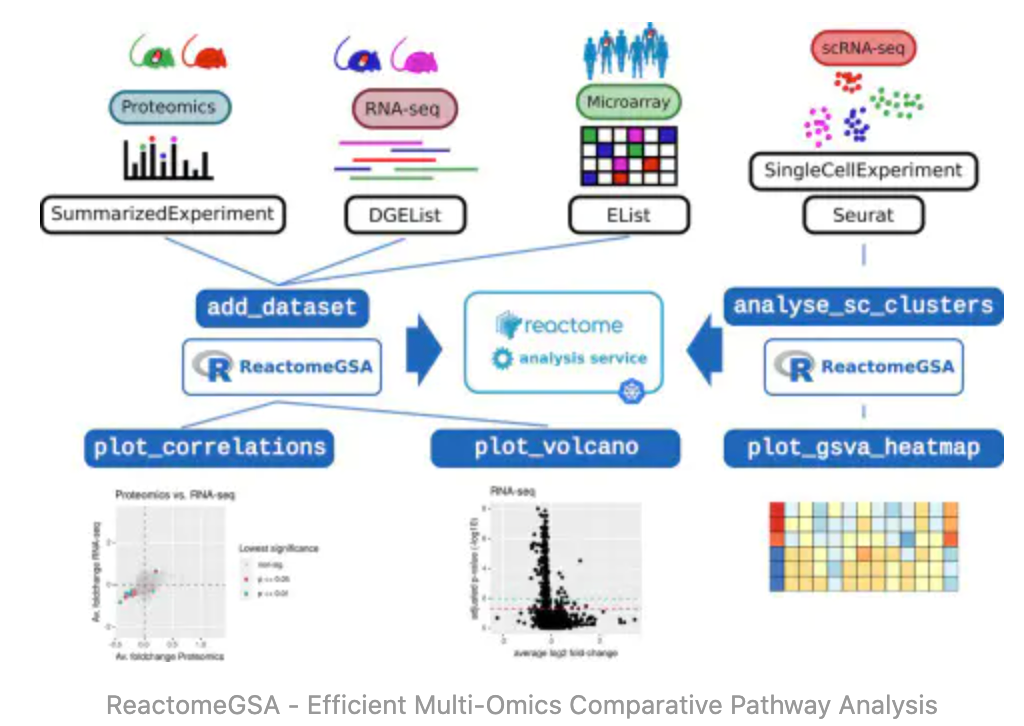
这里我们只看ReactomeGSA 处理单细胞的过程,其他过程可以自行探索。
if (!require(ReactomeGSA))
BiocManager::install("ReactomeGSA")
#> Loading required package: ReactomeGSA
# install the ReactomeGSA.data package for the example data
if (!require(ReactomeGSA)) BiocManager::install("ReactomeGSA.data")
library(ReactomeGSA.data)
library(ReactomeGSA)
#> Loading required package: limma
#> Loading required package: edgeR
#> Loading required package: Seurat
data(jerby_b_cells)
jerby_b_cells
An object of class Seurat
23686 features across 920 samples within 1 assay
Active assay: RNA (23686 features, 0 variable features)
> head(jerby_b_cells@meta.data)
orig.ident nCount_RNA nFeature_RNA
cy53_1_CD45_neg_H12_S384_comb cy53 50508 8124
Cy81_FNA_CD45_G01_S265_comb Cy81 292554 3301
cy94_cd45pos_F03_S159_comb cy94 240094 4246
cy79_p4_CD45_pos_PD1_pos_F03_S351_comb cy79 177665 3824
cy94_cd45pos_C05_S125_comb cy94 830771 7004
CY89A_CD45_POS_6_H05_S185_comb
输入一个Seurat对象,analyse_sc_clusters就可以完成Seuat的基本分析。
gsva_result <- analyse_sc_clusters(jerby_b_cells, verbose = TRUE)
Calculating average cluster expression...
Converting expression data to string... (This may take a moment)
Conversion complete
Submitting request to Reactome API...
Compressing request data...
Reactome Analysis submitted succesfully
Mapping identifiers...
Performing gene set analysis using ssGSEA
Analysing dataset 'Seurat' using ssGSEA
Retrieving result...
head(gsva_result@results$Seurat$fold_changes)
Identifier Cluster_1 Cluster_10 Cluster_11 Cluster_12 Cluster_13 Cluster_2 Cluster_3 Cluster_4 Cluster_5
1 RPS11 407.214286 613.350 278.096774 204.72 104.8750000 436.271930 294.0825688 426.179245 514.200000
2 ELMO2 9.707143 23.050 12.838710 8.24 9.0000000 26.289474 0.6697248 12.273585 14.288889
3 PNMA1 11.214286 0.900 8.387097 0.00 11.8333333 14.605263 1.9908257 16.339623 2.533333
4 MMP2 86.792857 0.000 454.419355 0.00 186.0833333 196.833333 0.0000000 155.632075 1032.944444
5 TMEM216 2.292857 12.400 6.516129 3.76 0.5416667 6.342105 5.3577982 6.103774 6.188889
6 ZHX3 18.392857 12.375 14.483871 8.44 3.5000000 17.289474 5.3119266 11.990566 18.444444
Cluster_6 Cluster_7 Cluster_8 Cluster_9
1 374.258824 120.7090909 705.537037 236.9148936
2 1.823529 7.1090909 54.277778 15.9148936
3 2.717647 1.8727273 6.037037 0.8085106
4 0.000000 0.3272727 1.148148 0.0212766
5 9.682353 1.2727273 9.703704 2.0851064
6 4.658824 1.0363636 22.462963 10.6808511
可以看到其实是以亚群为单位的ssGSEA分析,也启发我们在做单细胞富集分析的时候,如果计算量太大就可以考虑用亚群的平均表达量来。在此也反映,单细胞数据分析的基本单位是细胞亚群而不是单个细胞。
通路分析就一个函数
pathway_expression <- pathways(gsva_result)
> colnames(pathway_expression) <- gsub("\\.Seurat", "", colnames(pathway_expression))
> pathway_expression[1:3,]
Name Cluster_1 Cluster_10 Cluster_11 Cluster_12 Cluster_13 Cluster_2 Cluster_3
R-HSA-1059683 Interleukin-6 signaling 0.1067040 0.09940645 0.1509142 0.10414911 0.11368072 0.11979177 0.11156012
R-HSA-109606 Intrinsic Pathway for Apoptosis 0.1063122 0.10317826 0.1116539 0.10910644 0.12484602 0.10068176 0.10487716
R-HSA-109703 PKB-mediated events 0.1274642 0.05289859 0.1062885 0.09587804 0.07340752 0.08322147 0.08406187
Cluster_4 Cluster_5 Cluster_6 Cluster_7 Cluster_8 Cluster_9
R-HSA-1059683 0.11429424 0.1151514 0.09620555 0.11560454 0.13346414 0.10350569
R-HSA-109606 0.10839244 0.1021714 0.10272290 0.11024641 0.11423797 0.11243046
R-HSA-109703 0.05573774 0.0458425 0.12398024 0.07708516 0.07818166 0.01426709
找出有差异的通路
# find the maximum differently expressed pathway
max_difference <- do.call(rbind, apply(pathway_expression, 1, function(row) {
+ values <- as.numeric(row[2:length(row)])
+ return(data.frame(name = row[1], min = min(values), max = max(values)))
+ }))
max_difference$diff <- max_difference$max - max_difference$min
# sort based on the difference
max_difference <- max_difference[order(max_difference$diff, decreasing = T), ] head(max_difference)
name min max diff
R-HSA-350864 Regulation of thyroid hormone activity -0.4873008 0.3750639 0.8623647
R-HSA-8964540 Alanine metabolism -0.5061766 0.2556097 0.7617863
R-HSA-190374 FGFR1c and Klotho ligand binding and activation -0.3439743 0.4157764 0.7597507
R-HSA-140180 COX reactions -0.3460100 0.3727507 0.7187606
R-HSA-5263617 Metabolism of ingested MeSeO2H into MeSeH -0.1932855 0.4938234 0.6871089
R-HSA-9024909 BDNF activates NTRK2 (TRKB) signaling -0.3786589 0.3011768 0.6798357
感兴趣通路的可视化,可以看出哪些亚群在该通路比较高的或比较低的。当然,我们这里没有做细胞类型注释,所以你也看不出具体的生物学意义啦。这里绘制的一条通路是 Alanine metabolism(丙氨酸代谢),肌肉组织中的氨基酸经转氨基作用将氨基转给丙酮酸生成丙氨酸,经血液运输到肝脏,在肝脏中丙氨酸通过联合脱氨基作用生成丙酮酸和游离氨,可经糖异生作用生成葡萄糖,葡萄糖由血液运输到肌肉组织中沿糖分解途径再产生丙酮酸,后者再接受氨基生成丙氨酸,通过这循环使肌肉中的氨以无毒氨基酸形式运输到肝,同时肝也为肌肉提供了生成丙酮酸的葡萄糖。
library(tidyverse)
p1 <- plot_gsva_pathway(gsva_result, pathway_id = rownames(max_difference)[1])
p1$data %>% mutate(absmy = ifelse(expr>=0, "Z","Fy")) -> df
?element_blank
df %>% ggplot(aes(cluster_id, expr ,fill=absmy ))+
geom_bar(stat='identity') + theme_bw()+
theme(axis.text.x = element_text(angle =45,hjust = .9,size = 10,vjust = 0.9))+
ggtitle("Alanine metabolism") + theme(legend.position="none")

# Additional parameters are directly passed to gplots heatmap.2 function
plot_gsva_heatmap(gsva_result, max_pathways = 15, margins = c(6,20))
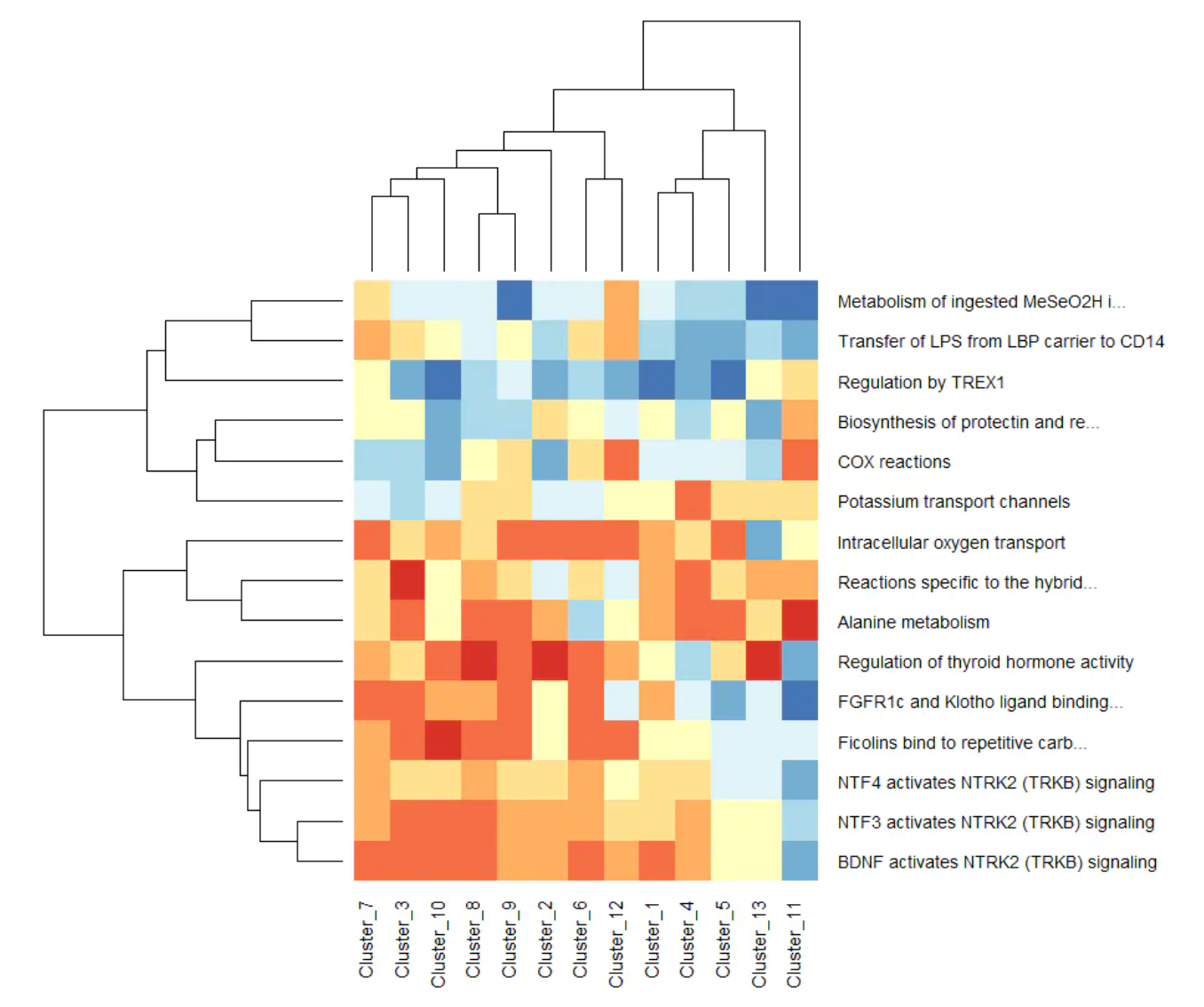
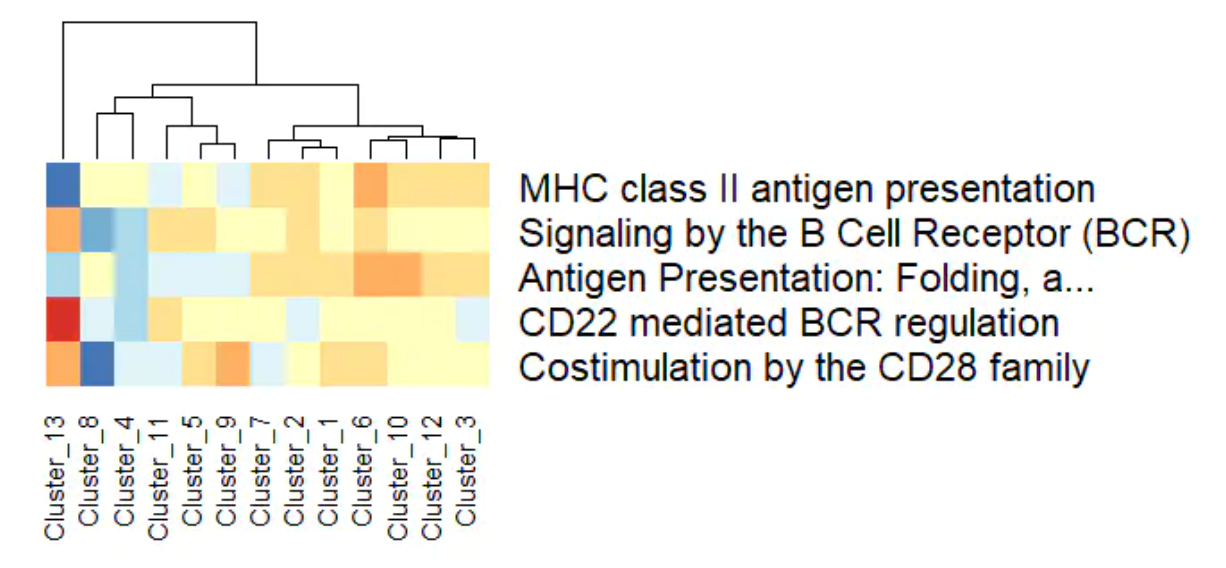
选择特定的通路
# limit to selected B cell related pathways
relevant_pathways <- c("R-HSA-983170", "R-HSA-388841", "R-HSA-2132295", "R-HSA-983705", "R-HSA-5690714")
plot_gsva_heatmap(gsva_result,
pathway_ids = relevant_pathways, # limit to these pathways
margins = c(6,30), # adapt the figure margins in heatmap.2
dendrogram = "col", # only plot column dendrogram
scale = "row", # scale for each pathway
key = FALSE, # don't display the color key
lwid=c(0.1,4)) # remove the white space on the left
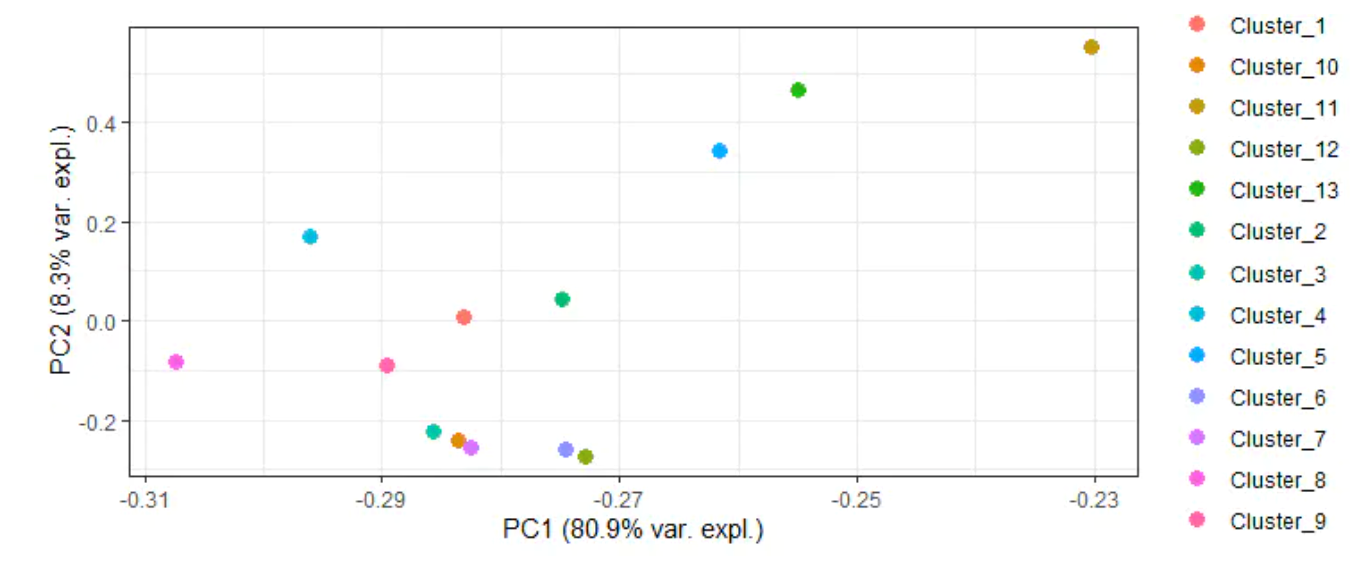
其实做完GSVA之后,我们相当于得到了一个关于通路的矩阵,也可以做降维聚类,如PCA,可以看到哪些亚群在pca空间上更为相近。
plot_gsva_pca(gsva_result)
 原始出处:
原始出处:Griss J, Viteri G, Sidiropoulos K, Nguyen V, Fabregat A, Hermjakob H. ReactomeGSA - Efficient Multi-Omics Comparative Pathway Analysis.Mol Cell Proteomics. 2020 Dec;19(12):2115-2125. doi: 10.1074/mcp.TIR120.002155
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#CTO#
56
#ReACT#
52