

编者按:肺动脉高压的分类系统分为五大主要组别,每个组别根据其病因和特征进行划分。近期的分类更新包括重新引入了“对钙通道阻滞剂(CCBs)长期反应者”作为特发性肺动脉高压(PAH)的亚组,更新了第2组和第3组PH的亚组分类,并将美托麦辛和卡非佐米列入了与PAH“确定相关”的药物名单中。这些更新反映了对PH理解的进步,并帮助临床医生更好地管理和治疗不同类型的PH患者。精确的分类对于制定个性化的治疗计划和改善患者预后至关重要。


肺动脉高压的临床分类
肺动脉高压(PH)的临床分类的主要目的是根据相似的病理生理机制、临床表现、血流动力学特征和治疗管理,对与PH相关的临床情况进行分类。2018年第六届世界肺动脉高压研讨会(WSPH)和2022年欧洲心脏病学学会/欧洲呼吸学会(ESC/ERS)指南为儿童和成人提供了简化且全面的分类版本,将PH分为五个亚组(表2)。我们建议保留PH临床分类的核心结构,但可能需要进行一些澄清和调整。以下我们提供了具体的评论,指出了可能存在的相关模糊区域和证据缺口,这些问题需要进一步研究。

常见和罕见形式的PH
“PH”一词定义的是一种血流动力学状态,而不是一种疾病实体。总体而言,PH是一种相对常见的状况,全球患病率约为1%。目前的PH分类将罕见的肺血管疾病归入第1组和第4组(PAH和CTEPH),而将与左心疾病、肺部疾病和/或缺氧相关的PH并发症归入第2组和第3组。
一项关于全球疾病负担的最新系统回顾发现,通过右心导管(RHC)确诊的PAH的平均患病率为每10万人3.7例。第2组和第3组PH是最常见的PH形式,约占全球PH病例的90-95%。在第2组和第3组PH中,大多数患者患有轻度至中度PH,肺血管参与有限。
一种替代分类可能仅专注于肺血管疾病。然而,在一些患者中,存在严重的PH及显著的肺血管参与,与基础疾病的严重程度不成比例,影响约1-10%的左心或肺部疾病患者。因此,完全排除这些疾病出肺血管疾病分类似乎具有挑战性。在第5组PH中,情况更加复杂,该组包括机制不明和/或多因素机制的PH,且有时伴有严重且特定的血管参与,例如肉芽肿病。因此,我们建议保留目前临床分类的架构。
此外,目前建议的临床分类旨在向非专业人士传递信息,因此强调列出所有可能的原因,以便在评估PH时加以考虑。
值得注意的是,在2022年ESC/ERS指南中,第2组、第3组和第4组的PH术语“由于(due to)PH”已更改为“与PH相关(assocaited with)”。我们支持这一改变,因为它强调了相关疾病的存在(如左心病、慢性呼吸系统疾病或慢性血栓栓塞性疾病)可能不足以引起PH,而是构成与复杂病理生理机制相关的风险因素。
伴随合并症的PAH
在目前的临床登记中,伴有心肺合并症的PAH患者的比例可能高达60-85%,即使在关键的PAH试验中,约50%的受试者也有心肺合并症。注册登记数据表明,PAH诊断时的年龄通常超过60岁,这增加了同时存在一般人群中常见的心肺合并症的可能性。我们承认,PAH患者可能会患有心肺合并症。同时,严重的心脏和肺部合并症的存在是将其分类为与左心或肺疾病相关的PH(第2组或第3组PH)的强烈指征。然而,当存在严重的毛细血管前参与,且仅有轻度或中度心肺合并症时,区分伴有合并症的PAH与第2组/第3组PH有时较为困难,这是当前知识中的一个空白。
在涉及心脏合并症的病例中,通常通过区分毛细血管前和毛细血管后的PH(即PAWP≤15 mmHg与>15 mmHg)来确定患者属于第1组还是第2组PH。值得注意的是,PAWP<15 mmHg并不能排除左心疾病的存在,且患者可能基于临床表现被分类为与左心疾病相关的PH。在PAWP值接近上限的患者中,全面的评估可能会揭示潜在的左心疾病。
在患有PH和慢性呼吸系统疾病的患者中,定义分类的阈值颇具挑战性。我们建议对这些患者进行多模式评估,包括肺功能测试(PFTs)、肺一氧化碳扩散能力(DLCO)测试和高分辨率CT(HRCT)。HRCT应特别在过去或目前吸烟且DLCO显著下降(<45%预测值)的患者中进行。对于肺功能异常显著或HRCT中有显著肺实质参与的患者,特别是肺纤维化和/或肺气肿,应优先分类为第3组PH。
总的来说,我们主张利用多模式的临床检查,包括详细的病史、超声心动图、磁共振成像(MRI)、肺功能测试和HRCT来对患有心肺合并症的患者进行适当的分类。我们认为不要仅依赖血流动力学测量或任何单一临床参数。我们不建议为伴有合并症的PAH患者引入一个子组。
具有多种PH机制的患者分类
患者可能会同时出现多种导致PH的疾病。此类例子很多,包括患有慢性阻塞性肺疾病(COPD)的患者,通常伴有显著的左心疾病和慢性血栓栓塞性疾病,或患有系统性硬化症的患者,这些患者不仅会发展为PAH,还经常伴有间质性肺疾病或肺静脉阻塞性疾病(PVOD)。这些复杂患者的主要分类应基于推定的PH主要原因。
结缔组织病(CTD)相关的肺动脉高压(PH)就是一个典型的分类挑战,PH可能由于多种重叠或不同的机制引发,并且发生在CTD相关的合并症背景下。这不仅对术语和分类很重要,还会影响PH的管理。最常见的CTD为系统性硬化症(SSc)、混合/重叠型CTD(MCTD)和系统性红斑狼疮(SLE)。研究最深入的CTD是SSc,大多数患者患有毛细血管前PH,原因是第1组PAH或与间质性肺疾病相关的第3组PH。然而,心脏受累,尤其是舒张功能障碍(射血分数保留的心力衰竭),是SSc-PH的一个重要因素。一些SSc患者中的其他相关机制包括PVOD和第4组血栓栓塞性PH。在患者中定义主要的PH机制是一个重大挑战,需进行专家PH评估和多学科管理,并进行适当分类,以便作出最佳治疗决策。在SLE和MCTD的病例中,优化基础疾病的治疗至关重要,因为这可能会显著改善PH。另一个需要考虑的是,PAH治疗药物,尤其是磷酸二酯酶-5抑制剂和波生坦,常被用于治疗SSc中的数字血管病变。需要进一步研究以了解这些药物如何影响PH的筛查、检测以及其发展和自然病程。
钙通道阻滞剂的长期应答者
大约12%的特发性肺动脉高压(PAH)患者或药物及毒素相关的PAH患者在急性血管反应性试验中会有阳性反应,而遗传性PAH患者的比例更小(<5%)。在这些病例中,急性血管反应性试验的阳性反应预示着患者可能对高剂量钙通道阻滞剂(CCBs)有长期应答。然而,对于其他形式的PAH及其他肺动脉高压(PH)群体,急性试验的结果可能具有误导性,真正的长期应答者非常罕见。即便有急性反应的患者中,少于三分之二的患者在仅使用CCBs治疗≥1年后表现出持续的临床和血流动力学改善。事实上,部分对CCBs有长期反应的患者可能逐渐失去这种反应,有时甚至在数年后病情会像特发性PAH患者一样进展。值得注意的是,最近发布的一项多中心研究表明,除了已确立的血管扩张应答标准外,急性血管反应性试验中的肺动脉顺应性、低风险状态以及早期随访时正常的N末端脑钠肽前体(NT-proBNP)水平,与长期应答及生存率密切相关。
在2018年第六次WSPH会议上,PAH(组1.5)中的“CCBs长期应答者”亚组被单独识别出来。将这些患者区分出来的理由在于其特定实体具有显著更好的预后、独特的管理方法和不同的病理生理机制,主要由血管收缩驱动,而非肺动脉重塑。然而,这一提议引发了关于分类随时间变化的质疑,至少在两种情况下:1)只有经过1年的随访后,患者才能被纳入此亚组;2)失去CCBs反应的患者,其病程与特发性PAH患者相似。
在2022年ESC/ERS PH指南中,“CCBs长期应答者”亚组被移出临床分类。取而代之的是,将特发性PAH(亚组1.1.1和1.1.2)中的“急性反应者”和“非反应者”进行区分。然而,这并未解决所有的空白和误解。临床分类的目的是将具有共同病理生理机制的PH归类。然而,急性反应者是CCBs长期应答者和需要使用PAH靶向药物管理的患者的混合体。此外,这是唯一一个仅由初始治疗策略(CCBs)而非病理生理机制定义的亚组。
我们主张重新引入“CCBs长期应答者”作为特发性PAH的一个亚组。我们承认,针对这一重要患者群体进行一致的统一分类具有挑战性。重新引入这一亚组强调了在特发性PAH、遗传性PAH和药物相关PAH中进行血管反应性测试的重要性(表2),并突出了对这些患者进行长期血流动力学和临床随访的重要性。
关于与发育基因突变相关的PH分类的挑战
最近一项关于PAH遗传咨询和检测的国际共识声明提出了对PAH易感基因的更新。最近发现的PAH易感基因(TBX4、SOX17、KDR)在肺血管、肺实质或支气管以及心脏发育中起作用,这导致了复杂的表型。这些表型有时包括先天性心脏病和严重的肺发育异常。
在最初被归类为先天性心脏病(CHD)相关PAH的儿童和成人中,尤其是具有SOX17突变的病例中,很多患者实际上是遗传性PAH患者。
在携带TBX4突变(小髌骨综合征)的患者中,表型差异很大,甚至同一家族中也是如此。PH可能与显著的肺发育异常相关,特别是在儿童中,或者在成人中表现为类似特发性PAH的特征,几乎没有或只有很少的肺发育异常。根据患者的情况,这可能被归类为两种不同的群体:遗传性PAH(组1 PH)或与发育障碍相关的PH(组3 PH)。
因此,这些患者的分类在很大程度上取决于基因检测的可用性和系统实施。对于CHD-PAH来说,影响较小,因为其管理类似于遗传性PAH。而对于与肺发育异常相关的PH,其分类为组1或组3 PH可能会导致不同的管理方法。这些情况下需要研究,以确定这些肺血管疾病的病理生理学最接近哪个组。
有关PAH风险药物的更新
WSPH和ERS/ESC指南中定期更新了明确或可能与PAH发展相关的药物分类。自第六次WSPH会议(2018年)以来,该分类已从三类(明确、可能和可能)演变为两类(明确和可能)。定义如下:“明确相关性”包括基于暴发、流行病学病例对照研究或大型多中心系列的数据;“可能相关性”则由多次病例系列或具有相似作用机制的药物病例提示。然而,这些标准并未考虑到用于评估药物与PAH因果关系的证据和标准的多样性。在药物相关PAH的特异性中,我们可能注意到1)根据药物不同,在PAH诊断前的暴露持续时间从几周到几年不等;2)某些药物在停药后已观察到疾病的回退,但并非所有药物都如此;3)仅在少数药物中已在临床前模型中再现PAH;4)药物流行病学和药物警戒研究产生了各种药物与PAH关联的估计值。
在第七次WSPH会议期间,我们提出了基于早期相同标准的药物相关PAH的更新分类。此外,我们考虑了其他因素,尤其是对可能与PAH相关的药物。这些因素包括停药后PH的可逆性、临床前实验数据的存在、典型PAH的组织病理学发现以及与明确相关PAH药物的共同机制。工作组强调了制定多模式和综合标准来评估药物相关PAH病因学证据水平的重要性(即判断药物与PAH相关性的合理性)。这些标准随后可用于计算因果关系评分,这可能成为新分类的基础。
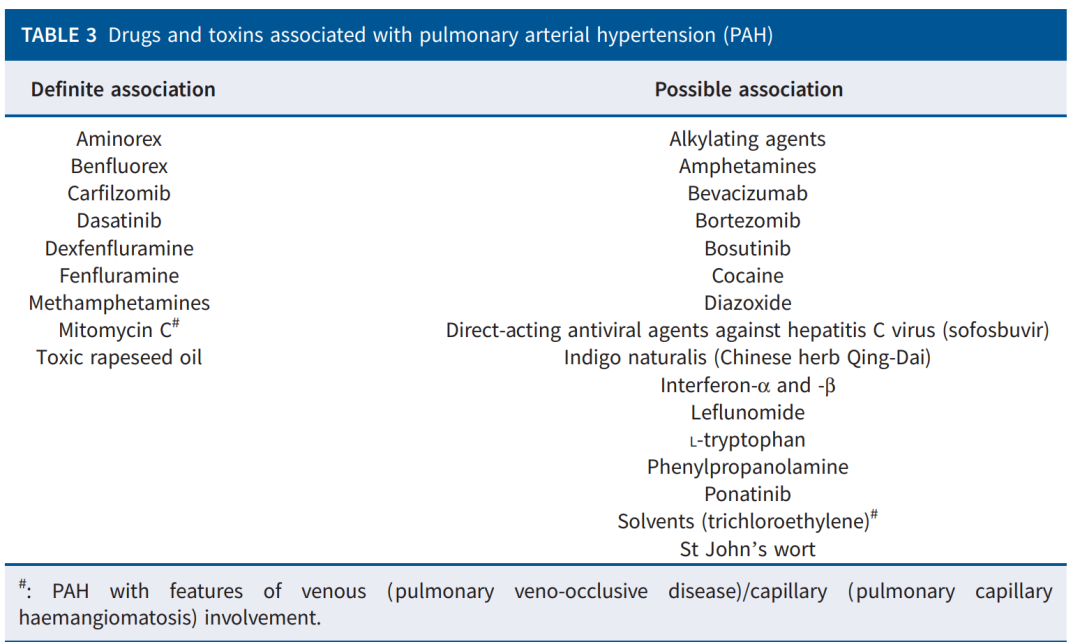
基于最近的数据,我们建议对2022年ESC/ERS PH指南中提供的与PAH相关的药物和毒素名单做出一些细微更改。丝裂霉素C和卡非佐米被添加到了“明确相关性”组。丝裂霉素C是一种生物还原烷化剂,已在系列病例和流行病学数据中报道与具有静脉/毛细血管(PVOD/肺毛细血管血管瘤病)特征的PAH相关。这一关联已通过人体组织病理学评估得到加强,并在动物模型中再现了该疾病的特征。由于数据有限,其他烷化剂仍然被保留在“可能相关性”组。在个别病例报告中,卡非佐米(一种蛋白酶体抑制剂)被报道与PAH相关。最近的一项研究使用了多种方法,包括国家PH登记处的分析、使用世界卫生组织全球数据库(VigiBase)进行的药物警戒不成比例分析以及随机对照试验的荟萃分析,显示了卡非佐米与PAH之间的显著关联。
我们建议将贝伐单抗作为“可能”与PAH相关的药物列入,同时将硼替佐米列为与PAH相关性较卡非佐米小的药物,但其潜在的共同病理生理机制相似。此外,我们建议将“靛玉红(中草药青黛)”替换为“青黛(中草药青黛)”,因为现有的关于这种草药及其与PAH相关性的病例报告是基于青黛的使用和购买,而不是靛玉红,靛玉红是青黛的药效成分之一。尚未确定青黛中与PAH相关的成分(表3)。
关于与HIV、门脉性肺动脉高压(PoPH)以及第5组肺动脉高压(PH)相关的PAH最新进展
近年来,多项研究通过大型数据库证实了之前关于较少研究的PAH形式的发现。例如,与没有HIV的退伍军人相比,感染HIV的退伍军人患PH的风险显著增加。在HIV患者中,CD4阳性细胞计数低和病毒载量高是预测PH的因素。此外,最近的一项基于人群的队列研究显示,HIV合并PH的患者存在较高的共病负担,并且PH的存在与更高的死亡率相关。在资源充足的国家,HIV相关PAH的发病率在过去十年有所下降,可能是由于HIV及其相关共病的管理得到了改善。在法国PH登记研究中,HIV相关的PAH占2008年新诊断PAH病例的约7%,而在2021年降至不足3%。而在资源匮乏国家中,HIV相关PAH的流行病学尚不明确。
对于门脉性肺动脉高压(PoPH)患者,美国一项大型国家队列研究发现,心脏指数是进行风险分层的关键血流动力学变量。另一项最新分析表明,PoPH患者的生存率与肝脏疾病的严重程度密切相关。接受肝移植的患者长期预后最好,而未接受肝移植的PoPH患者预后较差。
基于一个大型国际登记研究,已确定了对于伴有结节病的PH患者移植生存率下降的新临界值。mPAP ≥40 mmHg 和PVR ≥5 WU与不良预后相关。对于符合条件的患者,肺移植仍然是一个选择。伴有肺结节病的患者在肺移植后的生存率似乎与其他适应症的患者相似。影响生存率的主要因素是年龄较大和术前广泛的肺纤维化。
最近,一项前瞻性队列研究报告了成人肺朗格汉斯细胞组织细胞增多症的长期预后,提示如果能早期识别并治疗并发症,该病的预后可能比之前认为的更为乐观,10年生存率估计为93%。该队列研究中的PH累计发病率在5年和10年的随访中均低于5%。
最近,PH也被描述为常见变异性免疫缺陷(CVID)的可能并发症。CVID相关PH主要表现为毛细血管前的PH,可能由多种原因引起,包括门静脉高压、肺血管重塑、肺实质病变,偶尔也有纵隔淋巴结病引起的肺血管外压迫。
1型神经纤维瘤病患者也可能发展为PH。这类患者的PH机制可能是多因素的,包括间质性肺病和特异性肺血管病变。1型神经纤维瘤病相关的PH具有女性为主、DLCO低、功能和血流动力学损害严重的特征。
对于复杂先天性心脏病(CHD)患者,我们建议将其继续作为第5组PH的一个亚组,因为这些患者患有肺血管疾病,但根据当前标准不符合PAH的诊断。
第2组和第3组PH的新建议亚组
此前的PH指南根据肺功能测量将第3组PH(与肺部疾病和/或缺氧相关的PH)分类为“阻塞性肺疾病”、“限制性肺疾病”和“混合性限制/阻塞性模式的肺疾病”亚组。我们建议用临床诊断,如COPD、间质性肺病和伴有肺纤维化与肺气肿联合的疾病来替代这些分类,以识别与肺疾病和/或缺氧相关的PH患者(见表2)。这突出了患者临床表现的重要性,以及影像学(主要是胸部CT扫描)在肺部疾病表征中的作用,除了肺功能检查外,还需考虑这些影像学表现。此外,我们建议引入“非实质性限制性疾病”的术语,用于描述那些呼吸衰竭和限制并非直接与肺实质病变相关的患者。例如,肌肉骨骼疾病患者可能由于通气不足而发展为PH,可以归类于此亚组。
同样,在左心疾病相关PH的分类中,已区分出特定形式的心力衰竭和瓣膜性心脏病。
未来PH分类的可能性
当前的PH临床分类是基于流行病学和临床考虑,以根据其表现、血流动力学特征和治疗管理来对与PH相关的临床情况进行分类。与其他医学领域一样,未来的科学进展可能会基于病理生理学特征、基因背景以及个体的治疗反应,开发出更个性化的PH分类。这可能会允许在诊断和管理PH患者时采取更为定制化的方式,从而帮助克服当前临床分类结构的相对僵化所带来的限制。
参考文献:
Definition, classification and diagnosis of pulmonary hypertension; Gabor Kovacs, Sonja Bartolome, Christopher P. Denton, Michael A. Gatzoulis, Sue Gu, Dinesh Khanna, David Badesch, David Montani; European Respiratory Journal; Jan 2024, 2401324; DOI: 10.1183/13993003.01324-2024
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#肺动脉高压# #分类#
70