中国糖原累积性肌病诊治指南(三)
2016-07-28 《中华神经科杂志》2016年第1期 《中华神经科杂志》2016年第1期

Ⅳ型糖原累积病(glycogen storage disease type Ⅳ,GSD Ⅳ)GSD Ⅳ型又称为糖原分支酶缺乏症(glycogen branching enzyme deficiency),还可称为Anderson病(Andersen disease)。GSD Ⅳ型是一种罕见的常染色体隐性遗传的代谢性疾病,由位于3p12上的GBE1基因突变而致病,1,4-α-葡聚糖分支酶(1,4-α
Ⅳ型糖原累积病(glycogen storage disease type Ⅳ,GSD Ⅳ)
GSD Ⅳ型又称为糖原分支酶缺乏症(glycogen branching enzyme deficiency),还可称为Anderson病(Andersen disease)。
GSD Ⅳ型是一种罕见的常染色体隐性遗传的代谢性疾病,由位于3p12上的GBE1基因突变而致病,1,4-α-葡聚糖分支酶(1,4-α-glucan branching enzyme,GBE1)的作用是在糖原合成的最后一步将α-1,4糖苷键变为α-1,6糖苷键。分支酶的缺乏,导致糖原合成障碍,支链淀粉样多糖在组织细胞中聚集,包括骨骼肌、心肌、肝脏、脑、周围神经等。
一、临床表现
GSD Ⅳ型十分少见,国内仅有个别病例报告。临床异质性大,骨骼肌受累明显,可分为3个类型。
婴儿型:出生后即伴有严重低张力、肌肉萎缩、关节挛缩、神经损害、肝硬化、肝衰竭,婴儿期死亡。最严重的表现为胎儿运动不能伴畸形,新生儿期死亡。
儿童型:以肝病为主的患儿,出生时无明显异常,1岁左右出现渐进性肝、脾肿大,肝硬化,门脉高压,肝功能衰竭,可伴有肌张力降低、肌无力和心肌病,多在3~5岁死亡。以肌病为主的患儿,多在10岁前发病,表现为运动能力下降,四肢无力,可伴有肌病面容,肌肉萎缩,呼吸困难。常伴有心肌病,个别可出现心源性猝死、非进行性肝肿大,后期可能出现肝硬化和脾肿大。
成人型:可以骨骼肌受累为主,表现四肢近端为主的肌无力,下肢比上肢严重,可伴有肥厚性或扩张性心肌病,后期可出现肝硬化和脾肿大。也可以中枢和周围神经受累为主,称为成人葡聚糖小体病(adult polyglucosan body disease,APBD),临床表现多样,主要表现为运动功能异常、肌肉无力及萎缩、神经性膀胱、周围神经病、共济失调、痴呆。病程多为进展性,个别报道呈波动性病程。神经影像学显示脑白质多发异常信号、脑萎缩[44,45,46]。
二、实验室检查
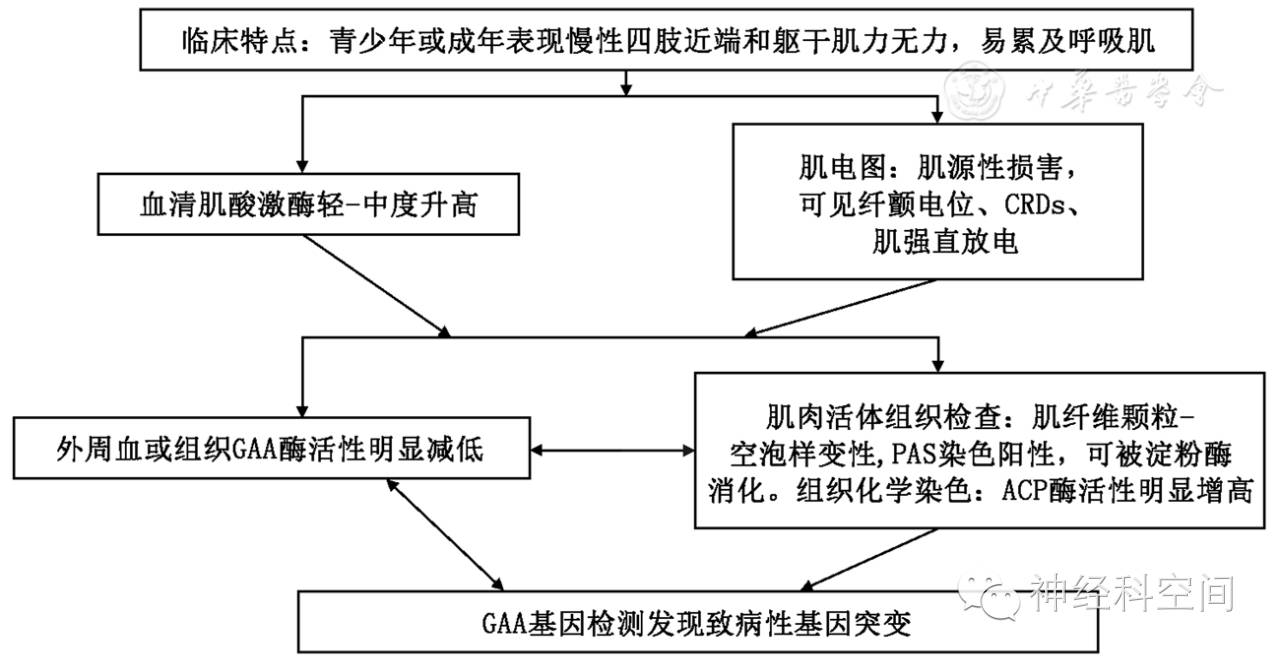
1.血清肌酸激酶正常或轻度升高。
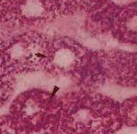
2.肌肉活检:以肌纤维内葡聚糖小体为病理特征,HE染色显示为肌纤维内嗜碱性异染物质,PAS染色阳性,且不能被淀粉酶完全消化[44]。电镜下观察除肌原纤维变性以外,可见无膜包裹的结构异常的糖原颗粒聚集,部分呈有分支的细线样。其他组织细胞中也可见到葡聚糖小体,包括周围神经神经轴索、脑组织、肝脏、心肌等[47]。
3.GBE酶活性检测:肝脏、肌肉、成纤维细胞或白细胞中分支酶活性明显降低或缺失。
4.GBE1基因检测:已报道的突变有20多种,包括纯合突变或复合杂合突变。
三、诊断依据
1.临床特点:不同程度的肝脾肿大、心肌病、慢性肌病,可累及中枢和周围神经,呈缓慢进行性加重。
2.肌肉活检:肌纤维内多发不能被淀粉酶完全消化的PAS阳性的包含物(葡聚糖小体)。电镜下可见无包膜的细丝和颗粒样结构聚集。肝细胞、神经细胞、周围神经轴索内也可见到葡聚糖小体。
3.GBE1基因检测发现致病性基因突变。不确定的患者,可以做分支酶活性测定。
四、鉴别诊断
临床上需要与先天性肌营养不良、脊肌萎缩症、先天性肌病、肌营养不良、肌原纤维肌病、GSDⅡ型、脑白质营养不良及其他遗传代谢性疾病相鉴别。病理显示的细胞内葡聚糖小体还可见于GSDⅦ型、Lafora病,健康老年人也可在个别肌间神经轴突内存在葡聚糖。
五、治疗
1.尚无酶替代治疗或其他药物治疗。
2.肝移植可以延长以肝硬化为主的患者的寿命,但总体上不能缓解其他组织器官的损害。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#诊治指南#
47
#肌病#
59