Menkes病:症状体征、病因、流行病学、诊断和治疗
2022-08-24 MedSci原创 MedSci原创

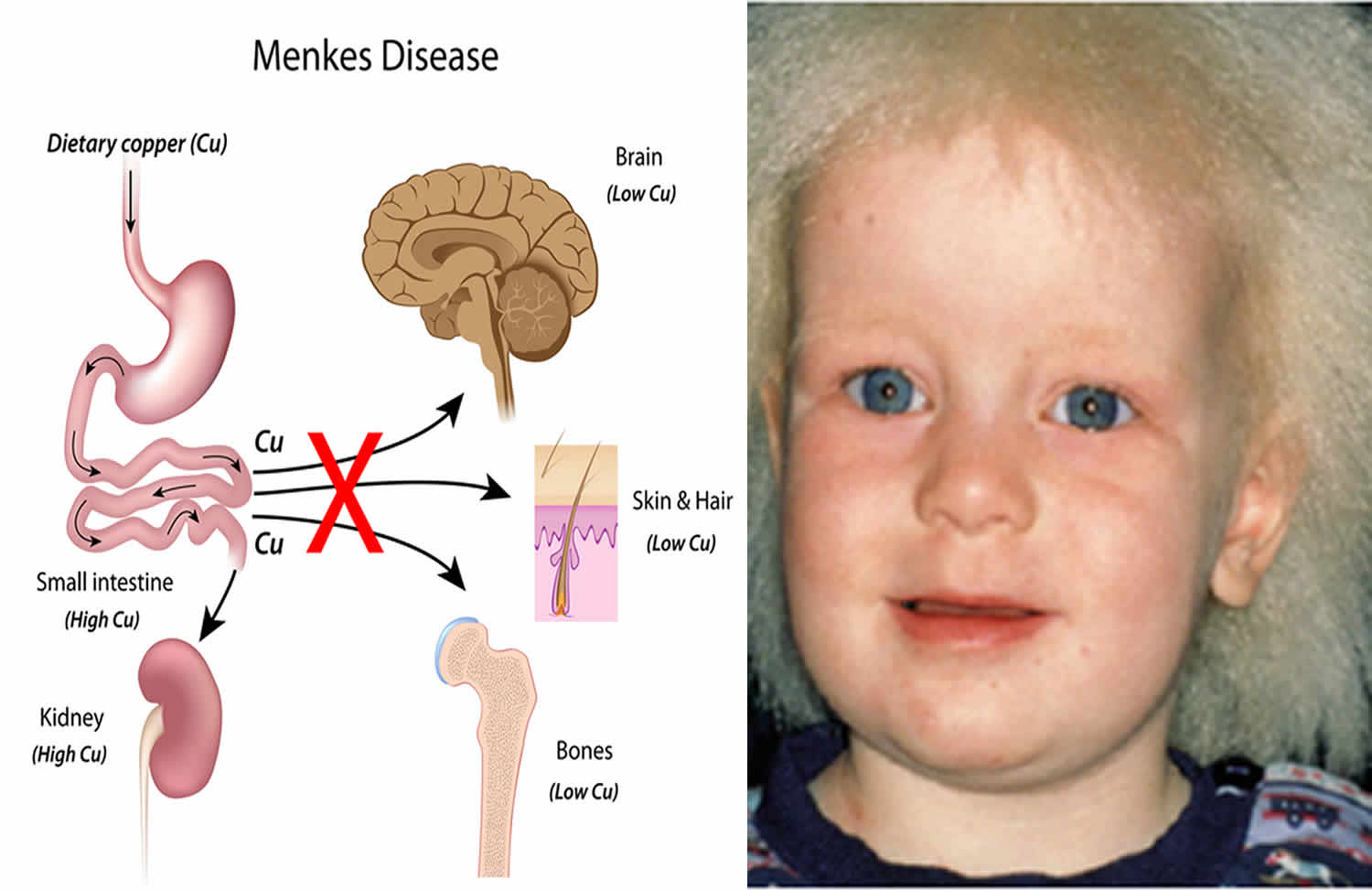
Menkes是指门克斯病,门克斯氏综合征,也叫做卷发综合征、毛发灰指营养不良等,为性连隐性遗传,是一种罕见的先天性铜代谢异常疾病。是由于铜在肠黏膜吸收以后,从黏膜细胞向血液转动过程的障碍,体内铜酶活性
Menkes是指门克斯病,门克斯氏综合征,也叫做卷发综合征、毛发灰指营养不良等,为性连隐性遗传,是一种罕见的先天性铜代谢异常疾病。是由于铜在肠黏膜吸收以后,从黏膜细胞向血液转动过程的障碍,体内铜酶活性降低,引起机体发育和功能障碍。门克斯病(Menkes disease)是1995年经全国科学技术名词审定委员会审定发布的医学名词 。
Menkes为X连锁隐性遗传病,男性发病,女性为携带者,1/3病例被确定为基因突变。孩子在出生时比较正常,多数在2-4个月开始出现严重的神经系统退化表现,一般是在3岁之前死亡,也有一部分患者能够生存到20岁以上。
发病以后由于哺乳不良,可导致身高和体重停止增长,孩子的智力发育也显著延迟,自发运动减少,肌张力降低或者是增高,腱反射亢进和四肢软瘫等锥体束征。Menkes主要的特征性表现为毛发呈连珠毛、结节性裂毛,且头发易折断,总体发病率较低。
一、一般介绍
门克斯病 (MD) 是一种遗传性 X 连锁隐性遗传疾病,会影响身体的许多系统。受影响的婴儿通常早产,可能有非特异性症状,如体温过低、低血糖和黄疸时间延长。一个明显而具体的体征是“钢”或“卷曲”的头发,通常在几个月大的时候就长出来了。门克斯病还与癫痫发作、发育迟缓、发育迟缓、体温不稳定和智力障碍有关。
门克斯病是由负责铜在全身运输的 ATP7A 基因突变引起的。身体使用铜作为辅助因子来激活某些酶以执行某些功能。当这些酶不能正常工作时,随着时间的推移,与控制头发、大脑、骨骼、肝脏和动脉发育的铜依赖性酶的功能有关的严重和致命的影响可能会随之而来。由 ATP7A 基因突变引起但症状较轻的 MD 变体包括轻度 Menkes 病和枕角综合征。
目前还没有完全治愈门克斯病的方法,但如果在出生后大约 28 天内及早开始,使用肠外组氨酸铜 (CuHis) 治疗可以提高存活率并减轻神经系统症状。
二、症状与体征
门克斯病的特征是皮肤干燥和头发异常,头发通常易碎、缠结、稀疏、钢铁般或卷曲,颜色通常为白色、象牙色或灰色。受影响的婴儿也可能出现由血液中过多的胆红素(高胆红素血症)引起的黄色外观(黄疸)。低于正常体温(体温过低)也可能出现在新生儿期。疾病的正常、无症状阶段通常持续两到三个月。
大脑和认知异常是这种疾病的核心。可能会出现血栓(硬膜下血肿)和/或脑动脉破裂或血栓形成。也可能出现神经退行性影响,例如癫痫发作和生长发育延迟。骨密度降低(骨质疏松症)很常见,并可能导致骨折。硬膜下血肿和骨折的结合可能导致对虐待儿童的错误诊断。肺气肿、膀胱憩室、颈内静脉静脉扩张引起的颈部肿块和皮质盲也有报道。
枕角综合征 (OHS) 被认为是一种较轻的 MD,神经系统受累较轻,通常在 5-10 岁左右被诊断出来。
三、病因与病理机制
Menkes病是一种由ATP7A基因突变引起的X连锁遗传疾病。该基因负责产生调节体内铜水平的ATP酶。 Menkes 病患者的大脑和肝脏中的铜含量异常低,而肠道和肾脏中的铜含量过多。如果没有铜作为其结构和功能的关键元素,身体的铜依赖性酶的活性就会降低。例如,铜酶酪氨酸酶的活性降低会导致头发和皮肤的色素沉着减少。铜酶赖氨酸氧化酶活性降低导致结缔组织无法形成坚固的内血管壁。
X连锁遗传疾病是由X染色体上的非工作基因引起的疾病,并在男性中表现出来。在其中一条 X 染色体上存在非工作基因的女性是该疾病的携带者。携带者女性通常不会出现症状,因为女性有两条 X 染色体,只有一条携带非工作基因。然而,由于男性有一条从母亲那里遗传的 X 染色体,如果男性继承了含有非工作基因的 X 染色体,他就会患上这种疾病。
X 连锁疾病的女性携带者有 50% 的机会让儿子患上这种疾病,有 50% 的机会让儿子不受影响。每次怀孕都有 50% 的机会生出像他们一样无症状的“携带者”女儿,并且有 50% 的机会生出非携带者女儿。
如果患有 X 连锁疾病的男性能够繁殖,他会将非工作基因传递给他所有将成为携带者的女儿。男性不能将 X 连锁基因传递给他的儿子,因为男性总是将 Y 染色体而不是 X 染色体传递给男性后代。
四、流行病学
最近的研究表明,Menkes 病的发病率约为 35,000 名活产男性中的 1 名。 大多数确诊的婴儿是男性; 然而,MD 仍可能发生在女性身上,这与不寻常的遗传情况有关。
五、鉴别诊断
威尔逊病是一种铜代谢的遗传性疾病,其特征是在各种身体组织中储存过量的铜,特别是肝脏、大脑和眼睛的角膜。 这种疾病是进行性的,如果不及时治疗,可能会导致肝(肝)功能衰竭、溶血危象、中枢神经系统功能障碍和死亡。 早期诊断和治疗可以预防严重的长期残疾和危及生命的并发症。 治疗的目的是通过螯合疗法减少体内铜的积累量,然后维持正常的铜水平。详细见:肝豆状核变性:症状与体征、病因、流行病学、诊断与治疗
六、诊断
几个月大时出现易碎、缠结、稀疏、钢化或卷曲的头发提示 MD 的诊断。 血液检查显示低水平的血清铜和铜蓝蛋白支持诊断。 重要的是要注意,这些水平在其他健康的新生儿中通常较低。 一种新的诊断方法可以在铜缺乏影响大脑之前识别受影响的婴儿,包括测量血浆儿茶酚胺水平。 这可能是未来门克斯病新生儿筛查的基础。 ATP7A 基因突变的分子遗传学检测也可能被证明是一种基于人群的新生儿筛查的有效方法。
对于因临床和生化原因怀疑受到影响的婴儿,可通过商业途径获得 ATP7A 突变检测以确认诊断。 一旦在受影响的家庭成员中鉴定出特定的 ATP7A 基因变异,也可以进行携带者检测和产前诊断。
七、治疗
Menkes 病的早期(理想情况下在出生后 28 天内,纠正早产)治疗是必不可少的。 注射组氨酸铜 (CuHis) 是一种制备为冷冻干燥产品的新分子实体,已显示可增加血液中铜的浓度并改善一些患者的神经发育结果。 CuHis 在老年、有症状的门克斯病患者中的疗效程度尚不清楚。
建议对受影响儿童的父母和家人进行遗传咨询,以正确了解复发风险。
八、科学研究
一次性腺相关病毒基因治疗与皮下 CuHis 注射相结合,代表了门克斯病患者正在开发的一种有前途的治疗方法。
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Vairo FPE, Chwal BC, Perini S, Ferreira MAP, De freitas lopes AC, Saute JAM. A systematic review and evidence-based guideline for diagnosis and treatment of Menkes disease. Mol Genet Metab. 2019;126(1):6-13.
Kaler SG: The neurology of ATP7A copper transporter disease: emerging concepts and future trends. Nature Reviews Neurology 2011; 7:15-29.
Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet. 2010;18(5):511-8.
Kaler SG, Holmes CS, Goldstein DS, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605-14.
Shim H, Harris ZL. Genetic defects in copper metabolism. J Nutr. 2003;133(5 Suppl 1):1527S-31S.
Mercer JF, Llanos RM. Molecular and cellular aspects of copper transport in developing mammals. J Nutr. 2003;133(5 Suppl 1):1481S-84S.
Llanos RM, Mercer JF. The molecular basis of copper homeostasis copper-related disorders. DNA Cell Biol. 2002;21:259-70.
Andrews NC. Metal transporters and disease. Curr Opin Chem Biol. 2002;6:181-86.
Strausak D, Mercer JF, Dieter HH, et al. Copper in disorders with neurological symptoms: Alzheimer’s, Menkes, and Wilson diseases. Brain Res Bull. 2001;55:175-85.
Mercer JF. The molecular basis of copper-transport diseases. Trends Mol Med. 2001;7:64-69.
Harris ED. Cellular copper transport and metabolism. Annu Rev Nutr. 2000;20:291-310.
Menkes JH. Menkes disease and Wilson disease: two sides of the same copper coin. Part II: Wilson disease. Eur J Paediatr Neurol. 1999;3:245-53.
Menkes JH. Menkes disease and Wilson disease: two sides of the same copper coin. Part I: Menkes disease. Eur J Paediatr Neurol. 1999;3:145-58.
Kaler SG. Metabolic and molecular bases of Menkes disease and occipital horn syndrome. Pediatr Dev Pathol. 1998;1:85-98.
Kaler SG: Diagnosis and therapy of Menkes disease, a genetic form of copper
deficiency. Am J Clin Nutr. 1998; 67:S1029-1034.
Kaler SG: Menkes disease mutations and response to early copper histidine treatment. Nature Genetics 1996; 13: 21-22.
Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA: Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nature Genetics 1994; 8:195-202.
Kaler SG. ATP7A-Related Copper Transport Disorders. 2003 May 9 [Updated 2016 Aug 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1413/ Accessed March 3, 2020.
Online Mendelian Inheritance in Man (OMIM). Menkes Disease. Updated 12/20/2019. https://www.omim.org/entry/309400 Accessed March 3, 2020.
NINDS Menkes Disease Information Page. Reviewed 2019-03-27. https://www.ninds.nih.gov/Disorders/All-Disorders/Menkes-Disease-Information-Page Accessed March 3, 2020.
Chang CH. Menkes Disease.Medscape. Last Updated: December 10, 2019. 13pp.
www.emedicine.com/neuro/topic569.htm Accessed March 3, 2020.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#流行病#
89