解析!2016度药品审评报告:两升两降、四大亮点(附全文)
2017-03-17 佚名 E药经理人

自2015年8月国务院下发《关于改革药品医疗器械审评审批制度的意见》(44号文)之后,我国开启了药品审评审批制度上全方位的改革,而CFDA药品审评中心的工作也更受瞩目。
3月17日,CFDA药品审评中心发布2016年度药品审评报告。自2015年8月国务院下发《关于改革药品医疗器械审评审批制度的意见》(44号文)之后,我国开启了药品审评审批制度上全方位的改革,而CFDA药品审评中心的工作也更受瞩目。那么,2016年CDE的成绩单是怎样的?又有哪些亮点?E药经理人整理行业最关心的个关键:一是CDE过去一年的审评全貌,二是企业过去一年的申报全景。
两增两降
CDE的完成审评的情况主要表现为:
升:2016年药审中心完成审评数量共12068件,另有943件注册申请完成审评因申报资料缺陷等待申请人回复补充资料。与过去4年相比,完成审评的注册申请数量创下新高。其中与2015年相比,提高了26%。
降:排队等待审评的注册申请已由2015年9月高峰时的近22000件降至近8200件,创4年来新低(基本消除了注册积压)。
化药:
升:化药新药完成审评529件,与2015年相比,提高了81%
升:进口药完成审评257件,与2015年相比,提高了49%。
年份 完成审评总数 新药 仿制药 进口再注册
2013 632 42 36 0
2014 647 20 102 5
2015 544 60 83 4
2016 1362 101 20 19
化学药方面,新药上市申请完成审评690件,创下4年来新高;而仿制药申请完成审评3655件,与2015年相比基本持平。
创新药(NDA) 仿制药(ANDA) 化药新药 化药仿制药 化药进口药
2013 282 621 212 585 47
2014 283 689 231 587 84
2015 390 3668 292 3585 172
2016 690 3655 529 2973 257
中药领域,完成审评1362件,与2015年544件相比,提高了150%,另有106件中药注册申请完成审评因申报资料缺陷等待申请人回复补充资料。其中,中药IND申请和NDA数量较往年均有所提高,较2015年分别提高了161%和68%。
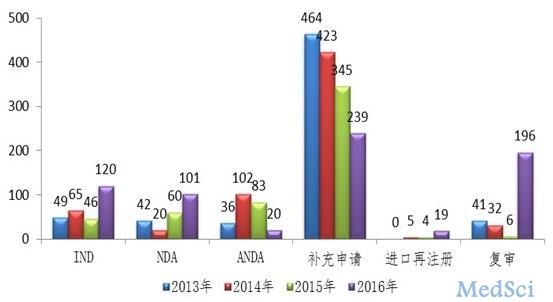
图:2016年完成审评送局的中药各类注册申请数量与前三年比较
生物制品上,2016年,药审中心完成审评并呈送总局审批的生物制品注册申请共646件,另有完成审评因申报资料缺陷等待申请人回复补充资料注册申请108件。
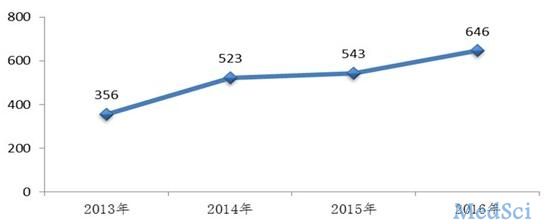
图:2016年完成审评送局的生物制品注册申请数量与前三年比较
企业申请注册情况主要表现为:
2016年,药审中心全年接收新注册申请3779件,较2015年下降了54%,且中药、化药和生物制品的注册申请接收量均有所下降,其中化药接收量下降幅度最大,降幅达57%。
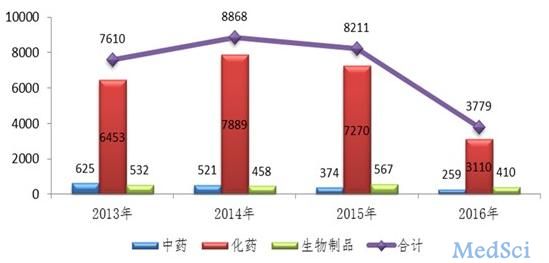
图:2016年注册申请接收情况与前三年比较
与前三年相比,2016年化药各类注册申请接收量整体呈下降趋势,其中验证性临床试验申请和ANDA受新注册分类实施影响接收量下降幅度最大,较2015年分别下降了80%和69%。但是化药创新药注册申请240件,较2015年增长了18%。
2016年新分类化药注册申请接收情况

四大亮点
2016年,药品审评工作取得了不错的进展,较为突出的几大亮点包括优先审评、生物等效性(BE)备案以及CDE审评力量建设等方面。
亮点一:优先审评,受益的有哪些品种?
自CFDA发布《关于解决药品注册申请积压实行优先审评审批的意见》以来,CDE已经共将12批193件注册申请纳入优先审评程序,其中中药注册申请2件、化药注册申请169件、生物制品注册申请22件。其中具有明显临床价值的新药注册申请共85件,占44%。
谁踏上了优先审评的快车道?根据报告,一批具有明显临床价值的创新药、临床急需药、专利过期药和国内首仿药完成审评并建议批准上市。这些产品分别是:

亮点二:BE备案:一致性评价品种16个备案
2015年底,化学药生物等效性试验开始由审批制改为备案管理。根据报告,2016年新申报化学仿制药BE备案品种41个,仿制药一致性评价BE备案品种16个。
从BE备案的数字来看,大部分还是新药品种,仿制药BE备案品种仅16个,也可看出当前大部分企业的仿制药一致性评价工作还处在前期研究阶段。
亮点三:首席科学家上任,审评人员扩至600人
2016年,CDE首次设立首席科学家岗位,并引进FDA资深审评专家何如意博士担任首席科学家。同时将审评中心人员扩增近600人。报告显示,CDE还创新人员培训模式,增加生产现场实训内容;制定药审中心《临床兼职审评员管理办法》等制度;积极探索项目聘用等灵活用人方式;进一步扩大药审中心在技术审评方面与社会的合作,与西苑医院、北京医院、清华大学医学院签署合作协议等。
亮点四:2016年CDE的审评大招
过去一年,CDE为提高审评效率和质量,可谓是用了洪荒之力。采用了加强审评项目管理、细化审评序列、强化时限管理,成立专项小组、增加审评人员、授权分级签发,制修订审评要点、规范技术要求,发挥省局挂职团队力量等“术法”。
突出的亮点在于,项目管理人制度与适应症团队审评制度。报告显示,CDE建立了近20人的项目管理人队伍,对内管理项目,制定工作计划,协调审评任务,服务于适应症审评团队;对外联系和服务申请人,组织申请人与审评人员沟通交流会议等,提高审评业务管理的专业性,使审评人员专心于技术审评,避免审评人员与申请人的私下接触,建立起廉政的“防火墙”。
其次是适应症团队审评制度初步形成。以肿瘤适应症团队为试点,以临床疗效为核心,临床、药学、药理毒理、统计等多专业审评人员与项目管理人员共同组成审评团队,实现了多专业审评、综合评价与集体决策。切实加强各审评专业间的沟通衔接,强化专业学科建设,明确主审人和审评员权责,依据风险大小授权不同层级人员签发,提高审评的科学性和规范性。
此外,在沟通交流制度上初见成效,创新药研发的沟通交流会122次。
2017年药品审评的核心关注点
总结过去,才能更好展望未来。最后来看看CDE在审评过程中发现了哪些主要问题?2017年的工作重点又是什么?
报告数字显示,2016年,各类注册申请的审评结论为建议不批准的共计2139件。各类注册申请第1轮审评结论为补充资料的共计1654件。不批准或要求补充资料的注册申请的原因主要有以下几个方面:
创新药:IND前期的安全性研究不够充分或研究数据可靠性不足,临床方案中对受试者风险管控措施不足或整体设计欠完善;NDA临床试验规范性差,数据质量较差,临床试验结果可靠性不足。此外,NDA申报资料中生产工艺信息不够详细的问题也较为常见。
仿制药:仿制药药学工艺、质量标准等研究存在较大缺陷,稳定性研究存在不足;前期研究不够充分,与审评要求差距过大,导致申请人未能按期完成补充资料或在后期主动放弃补充资料。
进口药:进口上市注册的申报资料未提供国外上市的全部研究数据,关键信息缺失,资料翻译错误较多,可读性差;进口再注册申请未按批件要求完成上市后研究。
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#解析#
64
#药品审评#
61
尽快完善药品评审,增加更多的儿童用药及剂型
97