
脓毒症休克中间质的作用:理解流体动力学的关键?
2024-01-20 重症医学 重症医学 发表于陕西省

间质,虽然并不属于传统的循环概念,但实际上在维持液体稳态中作用关键。调节液体平衡是脓毒症休克的一个关键方面,众所周知,液体平衡与预后有关。调节毛细血管内外液体流动是理解脓毒症期间液体稳态的第一把钥匙。

摘要
背景
间质,虽然并不属于传统的循环概念,但实际上在维持液体稳态中作用关键。调节液体平衡是脓毒症休克的一个关键方面,众所周知,液体平衡与预后有关。调节毛细血管内外液体流动是理解脓毒症期间液体稳态的第一把钥匙。
主要内容
脓毒症休克期间,毛细血管通透性增加,过去普遍认为,毛细血管渗透性增加足以解释为何炎症期间毛细血管滤过增加。然而,血管内皮壁的另一侧,间质可能在推动毛细血管泄漏方面作用更大。事实上,间质的细胞外基质形成了嵌入胶原骨架的复杂凝胶状结构,且能通过降低间质细胞外基质的静水压直接将血管内液体吸引到间质中。因此,间质,不仅仅是一个长期以来认为的被动储液池,而且可能是脓毒症休克期间调节液体平衡的主要决定因素。然而,迄今为止,很大程度上忽视了间质在感染和脓毒症休克中的作用。对间质的全面了解可能有助于理解脓毒症休克的病理生理学。总体而言,我们已经确定了间质在脓毒症休克病理生理学中的五大潜在作用:1.增加水肿,影响器官功能和代谢产物扩散;2.调节间质压力,增加毛细血管内外液体流动;3.改变细胞外基质;4.分泌炎性介质;5.降低淋巴液流出。
结论:我们旨在综述文献并总结当前这些特定领域的知识,以及研究间质相关的方法论知识。
关键词:间质,脓毒症休克,微循环,毛细血管泄漏,细胞外基质
背景
间质通常定义为任何特定组织细胞间的真实间隙,但其本身也是一个有结构的组织。随着活体内实时显微观察的进步,我们现在能够描述大的具有厚厚的胶原束限制的充满液体的多边形解剖空间(图1),特别是当这些空间位于皮下和消化道粘膜下层时。间质约占20%的体重,主要视为一个被毛细血管壁隔开的、与血管区相对应的液体区。过去循环的概念中,间质不属于循环。然而,在心血管和淋巴系统间,通过维持整个身体内的连续液体循环,间质是维持内稳态(转运代谢底物、废物和免疫介质)的重要环节。血液和间质区间的持续流体转运由所谓的Starling力控制。在脓毒症和脓毒症休克期间,Starling力急剧变化所致的强烈毛细血管渗漏,是导致脓毒症休克早期低血容量的原因。弥漫性水肿,是脓毒症休克后期的常见特征,也直接与毛细血管渗漏有关,但由于水肿临床识别延迟,掩盖了这种关系。脓毒症引起的水肿被液体治疗加重,多数时称为液体过负荷。近年来,已确定液体过负荷为脓毒症休克患者致残和致死的主要预后因素。
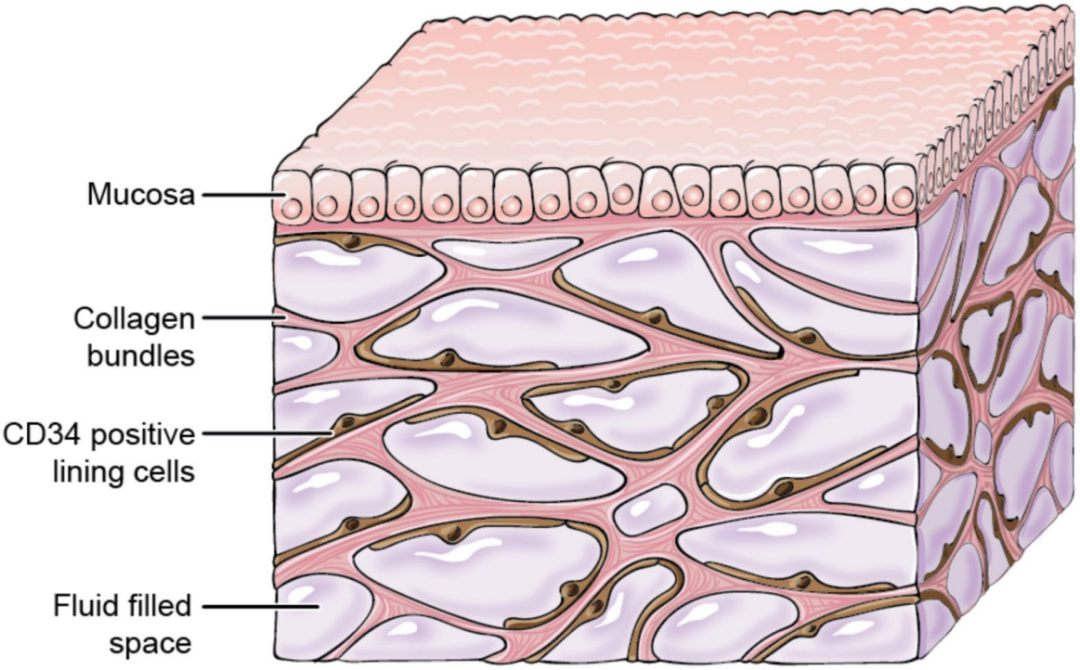
Mucosa粘膜、Collagen bundles 胶原纤维束、CD34 positive lining cells CD34阳性的粘膜细胞、Fluid filled space充满液体的空间
图1:间质的三维结构,空间示意图显示一侧排列着细胞的胶原纤维束网络,内部充满液体。
由于间质扩张,短短几天内,脓毒症休克患者体重就可能增加三分之一,因此应重视这个“第三空间”,重症医生也应全面理解间质。长期以来,人们只关注了血管腔室,一直忽视间质,将其视为被动的储备库。过去几十年里,生理学的重大进展,揭示了间质在急性炎症过程中(特别是在调节毛细血管过滤方面)发挥了重要作用。
总体而言,我们已经确定了间质在脓毒症休克病理生理学中的五大潜在作用(图2):1.增加水肿,影响器官功能和代谢产物扩散;2.调节间质压力,增加毛细血管内外液体流动;3.改变细胞外基质;4.分泌炎性介质;5.降低淋巴液流出。
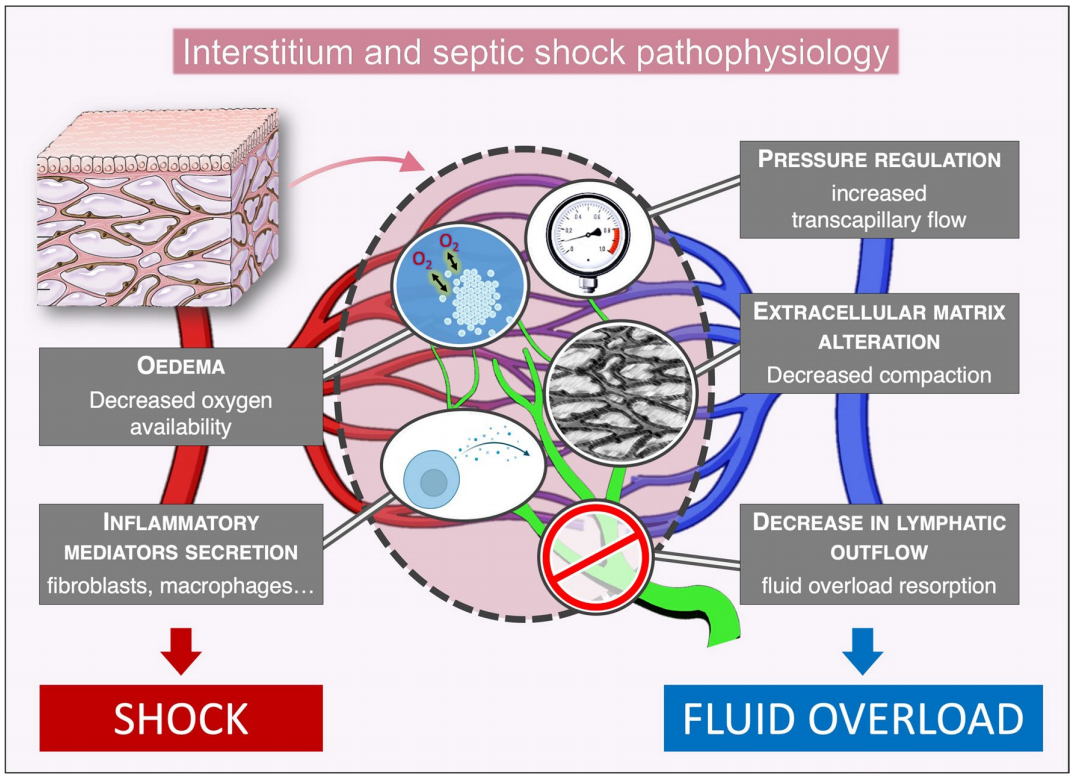
Interstitium and septic shock pathophysiology间质与脓毒症休克的病理生理学
PRESSURE REGULATION increased transcapillary flow 压力调节 增加的毛细血管内外流动
EXTRACELLULAR MATRIX ALTERATION Decreased compaction胞外基质改变 紧密性下降
OEDEMA Decreased oxygen availability 水肿 供氧减少
INFLAMMATORY MEDIATORS SECRETION fibroblasts, macrophages炎症介质分泌,成纤维细胞、巨噬细胞
DECREASE IN LYMPHATIC OUTFLOW fluid overload resorption淋巴流出减少,液体超负荷吸收
SHOCK休克
FLUID OVERLOAD 液体超负荷
图2:脓毒症病理生理学中可能的相互作用机制。毛细血管床周围的间质区室示意图(红色:动脉侧,蓝色:静脉侧)。淋巴管显示为绿色
为阐明间质在脓毒症休克病理生理学中的潜在作用,我们旨在回顾文献,总结文献上的当前知识,以及与研究间质相关的方法论方面。
间质解剖学和组织学
间质通常定义为一种分隔血管和细胞的液体隔室。最近的研究表明,无论是组织还是器官边界,间质空间都是连续的,微小物质可在结肠壁各层间或在皮下和筋膜间移动。所有器官都有间质组织,但其范围在不同的“母体”组织间差异很大,其结构和组成也可能有所不同,但外周血管周围间质与皮肤和腺体基底膜周围的松散结缔组织相似。尽管早就知晓间质的组成,但直到最近才观察到其三维结构(图1),这主要是由于体内显微镜技术的进步。通常,细胞外基质(ECM)由三维纤维胶原网支撑,其中胶原纤维形成厚厚的、宽20微米的束。微纤维-弹性蛋白框架也附着在胶原束上,将应力均匀地分布到间质结构中。基质由蛋白聚糖形成,不仅包括与核心蛋白共价连接的糖胺聚糖(GAGs),还包括游离的GAG(即透明质酸)。GAGs是由多个二糖单位组成的长链线性结构,极性很高并吸引水分,从而形成凝胶状结构。GAGs与ECM的胶原结构相互作用,但间质液中也有游离的GAGs。间质液本身是血浆的超滤物,其蛋白质浓度约为血浆的50%。间充质细胞覆盖在ECM外面并提供支持。
水肿:急性间质性水肿的发生和后果
局限性炎症时,为快速募集控制病原体所需的体液(例如,抗体,补体)和细胞(中性粒细胞,单核细胞)成分,局部的血液流动和通透性增加。脓毒症期间,全面过度激活先天免疫导致弥漫性内皮改变,引起大、小循环功能障碍。在毛细血管水平,内皮屏障改变可能是脓毒症最重要的后果,导致每层内皮壁都受损,从而使得毛细血管渗漏。VE-钙粘蛋白是内皮细胞间连接的主要组成部分。而由炎症细胞因子(TNF-α,IL-1β,IL-6和IL-10)引发的VE-钙粘蛋白内化(译者注:内化通常指的是细胞吸收或摄取某些物质(如蛋白质或更大的分子)的过程。在这里内化可能指的是VE-钙粘蛋白在炎症细胞因子的刺激下被细胞吸收的过程),足以破坏细胞间连接,从而增强血管通透性,这也受到其他因素的影响,如糖萼的改变。血管通透性增强经常认为是脓毒症休克期间驱动和维持毛细血管渗漏的必要充分条件。然而,应该记住,其他“力量”也正在起作用,并驱动与脓毒症相关的液体过载。
Starling定律
毛细血管滤过从而形成水肿的病理生理学,最初由Ernest Henry Starling在1896年描述。作为各种因子的函数,跨毛细血管过滤可用以下公式中表达:
![]()
在这里
Jv是每秒经内皮滤过的体积,Kf是膜的过滤系数,Pc是毛细血管静水压,Pi是间质静水压,πc是毛细血管胶体渗透压(COP),πi是间质胶体渗透压,σ是血浆蛋白的反射系数,ΔPf是净过滤压力。
毛细血管的静水压(Pc)从小动脉端的30-40 mmHg逐渐下降到静脉端的10-15 mmHg。血浆和间质渗透压分别约为28 mmHg和8 mmHg。正常的间质压力略负,在-1到-3 mmHg间。经典的Starling定律观点预测,由于毛细血管静脉端Pc和πi的下降,从间质吸收到血管腔上问液体吸收应发生在毛细血管的静脉端,这就一定程度上补偿了血管腔中滤出的体积。根据大量先前的实验数据,2010年Levick和Michel修订了这一经典观点,得出了“滤过是动态”的规则。事实上,他们表明,经典的斯特林公式中,πi可由糖萼下方COP(πg)替代,这是因为内皮屏障中蛋白质的反射元素实际上是糖萼纤维基质,其作用类似于超滤筛。由于细胞间隙中的高流速,间质蛋白无法通过糖萼下空间扩散回流,πg恒定低于πi(约为10%),而经典观点认为间质压力可忽略不计。这解释了稳态下毛细血管静脉端,尽管Pc降到πc以下,但仍未观察到滤过。然而,瞬时吸收仍可能发生,例如出血时,这是由于Pc急剧下降和πc增加。吸收也自然发生在肠粘膜和肾小管周围间质中。
水肿预防因子
因此,修订后的Starling定律指出,淋巴引流在稳态下等于毛细血管过滤。然后,水肿的形成是由于毛细血管过滤增加和/或淋巴流出降低之间的失衡。生理学中,保护机体免受水肿的机制已有广泛描述,特别是Guyton在这方面做出了非常重要和显著的工作。首先,增加的毛细血管滤过增加了间质容积,又通过稀释降低了间质渗透压,从而反过来减少了滤过。防止水肿形成的另一个保护因素是间质顺应性,这定义为在间质容积(ΔVi)增加一定量时,变化的间质压力(ΔPif),每种组织的间质顺应性都不一样。在脱水的基线状态和液体过度的初始阶段,容积-压力曲线是线性的,这保护了组织免受水肿(图3,实线)。当极度液体过度时,顺应性可能是无穷大的,并且在皮肤和肌肉间质中观察到容积-压力曲线的平台。皮下间质组织顺应性特别大,可积累大量的水肿。液体过载时,皮下间质占总容积的确切大小难以评估,但可能超过50%,因为稳态时,皮肤和肌肉已占据了总细胞外容积的三分之二。
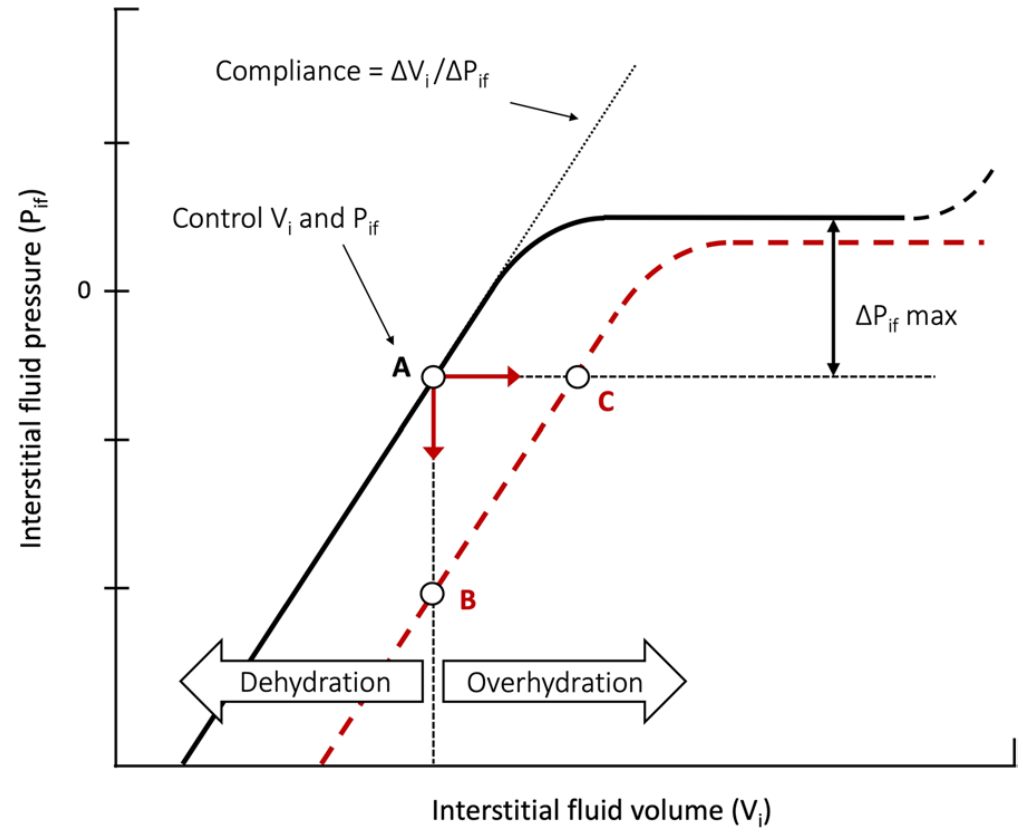
图3:稳态和炎症时的间质压力-容积曲线。正常状态下,间质容积-压力曲线的一般形状用实线(A)表示,实线含间质容积(Vi)和压力(Pif)的正常值。在脱水和过度水化的初始部分,顺应性(∆Vi /∆Pif)恒定。在获得Pif的最大升高(∆Pif max)后(反映了对过滤的最大对抗性压力),顺应性增加到无穷大(P/V曲线中的平台)。间质液体容积过度增加时,顺应性再次下降。炎症时,该曲线向红色虚线变化。如果过滤受阻,例如毛细血管血流中断,就会阻止间质容积增加,Pif会突然下降(B)。如果维持毛细血管流动,过滤率可能增加10-20倍,间质Vi可快速扩张,而间质压力变化很小(C)。
液体超负荷,脓毒症休克期间毛细血管渗漏的表现,同时也是预后因素
脓毒症休克的初期管理中,通常需要大量的复苏液体来补偿毛细血管的泄漏。水肿在所有组织(尤其是皮下组织)中积累,积累程度也反映了毛细血管泄漏的强度。近年来,已确定水肿(即,液体过负荷)为脓毒症休克患者的致残和致死的主要预后因素。然而,液体过负荷与不良预后间的关系性质尚未完全理解。首先,可以认为,急性期毛细血管泄漏强度与休克的严重程度直接相关。然而,另一方面,大多数发现这种关联的研究中,死亡率都根据初始休克严重程度做了调整(译者注:即在考虑了休克的初始严重程度后,毛细血管泄漏强度仍与死亡率相关)。此外,对脓毒症休克患者的观察性研究确定,液体负平衡的患者预后更好,这表明间质水肿本身可能会造成伤害。间质水肿增加了细胞间隙,长期以来一直认为细胞间隙是组织氧合的关键因素。实际上,氧分子(O2)是一种“疏水性”的、非极性分子,其在水中的溶解度和扩散性低,这使得其难以通过如间质液和细胞质等水性介质扩散。人们认为,氧经网状的脂质“疏水性通道”弥散,特别是在细胞内弥散时。因此,间质水肿与增加的弥散距离确实可能是脓毒症相关的氧摄取下降和代谢障碍的突出因素。间质水肿对预后效应的另一证明可在水肿对器官功能障碍的直接影响中找到,如水肿对急性呼吸窘迫综合症(ARDS)和急性肾损伤的直接影响。某些学者还描述了液体过负荷是腹腔间隔综合症的风险因素。
鉴于这些数据,减少液体过负荷已成为主要的治疗目标。目前,限制液体是旨在减少液体过负荷的主要可选治疗策略。2015年至2020年间发表的荟萃分析并未发现任何显著的益处,无论是全因死亡率还是如急性肾损伤或肺损伤等次要预后结果。2022年,Meyhoff等人报告了CLASSIC研究的结果,该研究也未发现限制性和自由性液体策略间的死亡率(或任何次要预后)有任何差异。2023年发表的CLOVERS试验证实了这些发现,没有死亡率差异,且限制输液组的升压药物使用增加,增加了不良事件但未达到统计学差异。总的来说,对液体平衡干预的效应较小,并不总具有统计学意义。这些试验的阴性结果强调了这个问题是复杂的,并暗示限制液体的方法可能不合适。
间质液评估:间质容量
水肿开始形成的时间非常早,但只在脓毒症休克后的24-48小时才明显,因为间质液约4升以下时,临床上无法检测到水肿。实际上,对脓毒症休克中液体超负荷的临床评估不足,缺乏敏感性。每日液体平衡监测是测量液体超负荷程度的最简单、最直接的方法。在过去一个脓毒症休克患者队列研究中,Boyd报告了到第4天的累积平均液体正平衡为+11升,更正的液体平衡与这些患者的死亡风险增加相关。为最准确测量血液腔室容积和跨毛细血管滤过率,需要注射一剂放射性碘标记的白蛋白,这样就能测量跨毛细血管逃逸率(TER)。该技术旨在评估脓毒症期间白蛋白输注的益处,但未能证明白蛋白输注能降低血管通透性。在一个使用同位素测量血容量和TER分析的重症患者(大多数为脓毒症休克)的随机研究中,44%的病例通过分析改变了液体管理,干预组的总体死亡率显著降低。这突显了评估重症患者液体状态的困难。通过晶体液输注引起的血液稀释,使用“容量动力学”方法,巧妙地探索了血管内和间质容积间的差异。该方法能预测健康志愿者和麻醉患者的血浆和间质中的液体分布,并且还证实了Guyton预测的“间质白蛋白洗脱”现象(译者注:“间质白蛋白冲洗”现象是指间质中的白蛋白被转移到血浆中。这可能是通过加速淋巴流动或通过某种其他机制募集白蛋白实现的。该现象最初是由著名的生理学家Arthur Guyton提出的,用来对抗毛细血管过滤增加时的周围水肿。该现象表明,尽管液体过载可能会导致间质水肿,但是,通过这种“间质白蛋白冲洗”机制,白蛋白和液体可以从间质空间返回到血管,从而帮助维持血管内的容积,然而这种机制在几个小时后可能会耗尽)。
炎症时的间质压(和跨毛细血管血流)调节
测量间质压
Arthur C. Guyton是测量间质压的先驱者,他在动物模型中通过植入穿孔胶囊开发了测量间质压的标准技术。在几周时间穿孔愈合和稳定后,就可测量压力。1980年代,有了许多测量间质压力的新方法。然后在动物模型中开发了各种皮下导管技术,使用充满液体的侧向导管或渗透性织物(wick)的注射针技术(译者注:这个技术的基本原理是将带有织物的针插入组织间隙,使织物与周围的间质液体接触。由于织物是渗透性的,液体可以通过织物渗透到内部。通过测量渗透到织物中的液体的压力,可以推断组织间隙的压力情况)。此后,大多数实验研究都使用了与自动反压系统连接的玻璃微管。最后,压力传感器的微型化使得能开发出可靠且准确的用于测量压力的导管,尽管它们的尺寸仍显著大于微管。
烧伤模型中发现的间质压力调节
如果升高Pif(间质压力)可防止水肿,那么降低Pif就足以增强毛细血管的过滤并促进水肿。健康志愿者中,对下肢施加负压会导致腿部间质压力几乎瞬间下降,液体滤过增加,腿部出现水肿。炎症期间,迅速形成的水肿不仅因内皮屏障破坏,还因间质压力的突然下降。实际上,烧伤时,明显的水肿会在几分钟内出现。间质容积必须至少增加一倍,水肿才会明显。因为间质液通常在12到24小时内更新一次,因此过滤率必须在短时间内增加数百倍以上,才能产生水肿。研究测量了实验性烧伤损伤期间Kf(毛细血管渗透系数)的变化,发现Kf增加了2到3倍。通过这种中度的Kf增加,计算出净过滤压力必须增加到200mmHg,才能解释观察到的过滤率。为验证该假设,Lund等人在大鼠的热烧伤实验模型中测量了间质压力。他们观察到,热伤后,皮内Pif从正常水平的-1mmHg降低到高度负值(-150mmHg)。因此,确认了净过滤压力增加的假设,主要是由于Pif的降低,而不是Pc(毛细血管压力)的增加。这项具有里程碑意义的研究,其结果随后被其他团队在类似的模型中证实,它首次证明了间质组织通过“吸引”在水肿中发挥了积极作用。
对其他局部和全身性炎症模型的拓展
“间质吸引”现象已在全身性炎症模型中得以再现,例如在通过静脉注射右旋糖酐引起的大鼠过敏反应中,皮下间质组织中的Pif降低了10 mmHg。同样,“脓毒性”炎症似乎也有相同效应。在实验性大鼠模型中,注射LPS降低了间质压力,并促使水肿形成和I-125标记的白蛋白渗出。在之前狗的内毒素性休克模型中,观察到类似的降低,间质压力立即降低了高达9 mmHg。
除了炎症,这种间质“吸引”现象的力量也在唾液腺中得到了展示,它使得蔬菜刺激后能产生唾液的爆发分泌,这时跨毛细血管流量增加十倍。这个生动的例证,说明“间质吸引”在日常生理功能中,有能力在有限的时间内从循环中募集大量液体。
有趣的是,研究发现肾上腺素的血管收缩和升压效应也会降低间质液压(Pif)。在他们的研究中,Border等学者探索了不同类型的休克和单独应用血管加压药对使用Guyton技术测量的间质液压的影响。如前所述,内毒素休克的特征是Pif的显著但短暂的降低(由于毛细血管滤过增加导致Vi增加,这解释了为何会恢复到休克前的Pif值)。然而,在出血性休克和单独应用儿茶酚胺的情况下,Pif也降低了,但降低是恒定的且与儿茶酚胺剂量相关。据作者的预测,这是由于血管加压药物减少了血管体积(从而相对增加Vi),这符合Starling原理。这些结果也可以通过由于极端血管收缩导致的毛细血管流量减少来解释。
细胞外基质和成纤维细胞的作用
因此,通过促进体液和细胞介质的转运,水肿并非偶然,而是代表了一种精细调节的机制,是局部免疫反应的必要组成。的确,如上所述,“间质吸引”现象会促进炎症性水肿。调节间质压力主要基于成纤维细胞和细胞外基质(ECM)间的相互作用。
成纤维细胞不仅与细胞外基质(ECM)相互作用,还在这个分子网络中调节间质压力。通过表达特定的整合素,尤其是通过其ß1亚基(例如,整合素α2ß1),成纤维细胞能结合到胶原纤维。整合素是包括2个亚基的跨膜蛋白,能双向粘附,能将细胞骨架粘附到ECM上。因此,通过与胶原的相互作用,ß1整合素亚基使持续的机械张力传递到细胞外基质,抵消在水化作用下糖胺聚糖(GAGs)自然扩张的趋势。因此,当对胶原纤维施加的压力快速释放时,可能会引起压力的突然下降(见图4)。
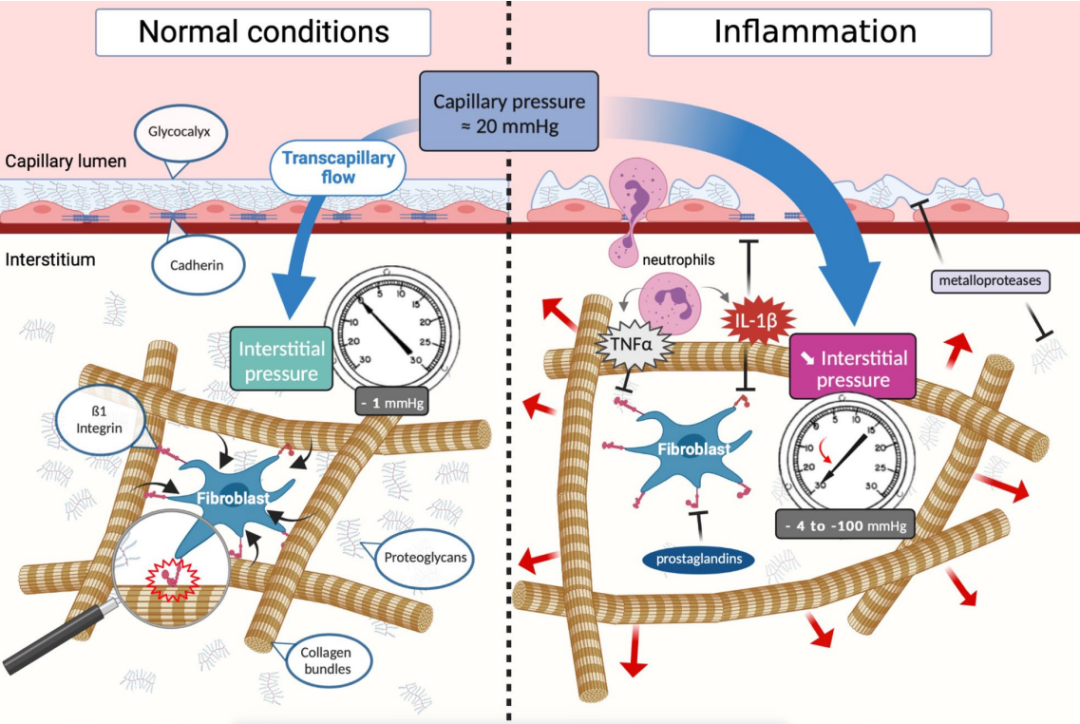
图4:微循环与间质细胞外基质在稳态(左图)和脓毒症期间(右图)的相互作用。稳态下,跨毛细血管流动受坚固的内皮屏障和稳定、轻微负压又紧密排列的间质调控(左图)。成纤维细胞对网状的胶原束施加恒定的张力。通过跨膜蛋白ß1整合素,成纤维细胞的细胞骨架与胶原IV相连。而炎症时(右图),炎症介质(尤其是IL-1ß)抑制了ß1整合素与胶原束结合,释放了束缚胶原网络的物理张力,导致间质压急剧降低。此外,各种由先天免疫细胞释放的蛋白酶不仅会改变糖萼,也会类似地改变细胞外基质。由于滤过压(毛细血管压力-间质压力)增加和细胞间粘附抑制和糖萼脱落导致的内皮通透性同时增加,使得跨毛细血管流动大量增加。。
体外实验中,通过评估成纤维细胞介导的悬浮胶原I型凝胶的收缩,证明了ß1整合素亚基在水肿形成中的作用。在该研究中,显示了成纤维细胞能够在24小时的培养期内将胶原凝胶收缩至初始凝胶体积的10%。这些发现得到了体内实验的验证,其中在大鼠皮肤中阻断ß1整合素粘附受体导致降低了间质压和水肿。炎症时,诸如IL-1β和TNF-α等促炎介质,通过作用于ß1整合素亚基,对抗该收缩,降低了间质压,进而形成水肿。血小板源性生长因子(PDGF)对与β1整合素相关的这种级联反应产生反向效应,通过收缩成纤维细胞和介导另一种整合素亚基(αVβ3)来增加间质压力。肿瘤中,间质扩大且间质压增加,PDGF抑制剂逆转了肿瘤诱导的间质压增加,并通过促进毛细血管过滤和抗肿瘤药物的局部扩散来改善抗肿瘤反应。
此外,细胞外基质(ECM)的糖胺聚糖(GAGs)也参与了间质容积的调节。GAGs带负电荷,特别能与细胞外液中最丰富的阳离子钠相互作用。根据GAGs的空间构象,其积累的钠不同,导致形成的间质体积也不同,有的地方会形成水肿和有的地方水肿会消退。脓毒症期间,由于醛固酮增多症,钠负荷增加,这一现象因ECM对钠的“结合力增强”而得到加强,总体上这可能阻碍水肿的消退,并干扰间质钠、巨噬细胞渗透感受器和VEGF诱导的淋巴管新生的复杂相互作用。
细胞外基质改变
细胞外基质(ECM)构建了间质空间的物理结构。炎症性功能性变化,导致了ECM不再紧密。此外,炎症还会直接改变ECM的结构性成分,这是由浸润的中性粒细胞被IL-1β和TNF-α激活后释放的强效催化酶引起的,如基质金属蛋白酶(MMPs)、肝素酶和透明质酸酶。脓毒症时,还发现了大量ECM的降解证据,从而提出了“全身性创伤”的概念。相对于对照组,在诊断脓毒性休克患者时,循环中交联的I型胶原肽段(ICTP,I型胶原降解的标志物)的水平要高五倍。这项研究中,胶原合成的标志物也增加了,尽管只有胶原III的前体(而非胶原I的前体)增加。相对于存活者,未存活患者的ICTP和胶原前体的水平也高。革兰氏阴性脓毒症的患者中,也发现ICTP水平升高,其他脓毒症患者的胶原III前体水平也增加。ARDS期间,胶原III前体也增加,并可能是纤维化进展的重要促进因素。
Koskela及其合作者们的一项小型描述性研究报告称,重症脓毒症早期,表皮层对粘连蛋白-332和IV型胶原蛋白的表达都下降,且在幸存者中持续了3个月。Hoffmann和他的合作者们报告称,重症脓毒症中基质金属蛋白酶及其抑制剂(MMP-9,TIMP-2和TIMP-1)浓度升高,而认为TIMP-1是预测重症脓毒症患者临床预后的有用生物标志物。另一项研究证实了TIMP1和MMP9在脓毒症期间的预后作用。
脓毒症时,细胞外基质(ECM)改变的后果目前尚不清楚。ECM改变可能会增加毛细血管过滤,因为ECM的物理结构对调节间质压力至关重要:胶原和蛋白聚糖的改变可能会破坏ECM的紧密结构。这得到了实验数据的支持:酶解反应增强了离体浸泡在等渗盐水中的松散结缔组织的肿胀,而正常情况下胶原纤维网络能限制这种肿胀。此外,提出维生素C对胶原的保护是其在烧伤模型中预防水肿效应的潜在机制。此外,ECM的改变在水肿的吸收过程中,即脓毒性休克液体管理的降级阶段,可能尤为重要。首先,与完整的ECM相比,改变的ECM可能丧失增加间质压力和启动淋巴引流压力梯度的能力。其次,间质中胶原的减少增加了间质蛋白含量,这是由Wiig等人描述的胶原纤维的“空间排斥”现象所致。事实上,对正常ECM而言,白蛋白和其他大分子的表观分布容积低于总的间质容积,因为由胶原网络限定的多个空间并不完全适合白蛋白和其他大分子分布。增加的间质蛋白质量可增加胶体渗透压,并直接损害液体转移,因为需要更高的淋巴流量(“洗脱”)来恢复间质液体体积。尽管临床数据尚缺乏,但恢复ECM结构可能是脓毒症休克患者实现液体负平衡的先决条件。
细胞外基质修复和适应
间质流动增加对ECM和驻留细胞有生物物理效应。机械应力及炎症可诱导成纤维细胞分化为肌纤维细胞,最终可能导致纤维化。研究发现,间质流动增加可通过上调MMP-1(基质金属蛋白酶-1)来诱导成纤维细胞运动,并通过TGF-ß依赖的机制驱动肌纤维细胞的分化和基质有序排列。在间质流动增加开始后的12-24小时内观察到ECM有序排列,而肌纤维细胞的分化则需要1-5天。值得注意的是,ECM的纤维垂直于间质流进行重新排列,降低了基质的传导性。这种重新排列的另一个值得注意的效应是剪切应力从细胞转移到基质纤维上。基质重新排列和成纤维细胞收缩也增加了ECM的硬化。总体而言,这些体外研究结果表明,持续增加的间质流可迅速导致ECM的硬化、重新排列和肌纤维细胞的分化,这些都是间质纤维化的标志。
目前尚不清楚是否会在持续炎症的重症患者中形成间质性纤维化,但这可能会导致愈合不良,并出现持续的液体超负荷状态,常见于更严重的患者中。
间质源性炎症(局部介质产生)
间质液评估:收集和分析间质液
鉴于间质的新兴作用,似乎有望从间质液中发现新的生物标志物。然而,获取间质液颇为困难。20世纪,为引流心力衰竭患者的水肿,曾皮下植入金属管(“Southey's tubes”)。目前已放弃这种有创性技术,但在ICU我们仍可观察到,明显水肿的患者中,皮下间质液很容易经皮肤裂口流出。在淋巴液流入淋巴结前行淋巴导管置管技术可能是获取“纯净”间质液的最佳途径,但有创,需要微创手术技能。微透析技术是为了确定间质组织的生化成分而开发的。目前主要用于监测重症脑损伤患者的缺血相关代谢变化。由于半透膜,微透析技术仅限于对离子和小代谢物的分析。而微灌注技术无需交换膜,因此能分析更广泛的分子,如细胞因子。在药理学研究中,尤其是局部使用的药物研究中,微灌注技术得到了广泛应用。
脓毒症和其他炎症性疾病中的间质探索
用于生物标志物分析的间质液比血液更难以获取,然而许多炎症标志物是局部产生的(,因此,如能够成功获取和分析间质液,可能会发现一些血液中无法检测到的重要生物标志物)。Olszewski等在类风湿性关节炎患者和对照受试者中进行了下肢淋巴管置管,并连续收集了72小时的流入淋巴结节前的淋巴液。对于测量的大多数促炎细胞因子(IL-1 β,TNF-α,IL-6,IL-8),发现淋巴液/血清浓度比大于1,就表明是局部产生。这些细胞因子也主要参与脓毒症的病理生理过程,并且大部分由巨噬细胞产生。此外,细胞因子浓度的个体间变异,或者由甲泼尼龙治疗引起的变异,能清晰地反应在淋巴液浓度上,而血清浓度并未显著变化。
Ikeoka等在九名重症脓毒症患者腹部皮下脂肪组织置入微灌注导管,发现IL-1β、IL-6和IL-8的浓度在皮下脂肪组织中高于血清,表明这些促炎介质由远离感染部位的皮下间质产生。此外,血压与IL-1β、IL-6和IL-8的皮下浓度负相关。内毒素血症大鼠模型中,使用皮肤离心法已证明了皮下间质产生IL-1,这表明在全身性炎症刺激期间皮下间质参与其中。免疫组织化学研究显示,产生这些细胞因子的细胞主要是间质成纤维细胞,还有表皮细胞和毛囊。在炎症性水肿形成中,这些学者还展示了IL-1β和TNF-α有浓度依赖性的直接机械作用,并报告了将内毒素血症中类似浓度的细胞因子应用于皮下组织后,间质压立即下降。在心脏手术患者的皮下脂肪组织中,也证明了远处间质产生细胞因子,间质液中IL-6增加,其源自具有核因子-κB调控基因激活的脂肪细胞。间质产生的细胞因子也可能也起源于Benias等学者在上皮下间质显微研究中观察到的一群有CD34+(一种通用的髓系细胞标记物,也存在于某些间质细胞表面)的间质成纤维细胞,实际上,当体外用肿瘤坏死因子(TNF)刺激时,有CD34的成纤维细胞分泌大量的IL-6、CXCL12和CCL2,这表明炎症期间它们有募集单核细胞的作用。
改变淋巴液流出
淋巴系统在循环中作用关键。稳态下,流入淋巴结前的淋巴液流量等于毛细血管过滤量,估计每天通过始于末端封闭的间质淋巴系统重吸收8-12升淋巴液。内皮细胞间的连接就像间质液的单向阀,引导间质液流入淋巴节前的收集系统。淋巴管壁中包裹的平滑肌细胞的节律性收缩以及单向阀,维持淋巴系统内的压力低于周围的大气压。与血管一样,淋巴管也对炎症介质做出反应。因此,经典的扩血管介质(如一氧化氮、前列腺素、组胺)与促炎细胞因子会下调淋巴管收缩。淋巴引流对抗原和树突状细胞转运到淋巴结非常重要。这解释了为什么局部炎症期间能观察到强烈的CD11+巨噬细胞诱导的淋巴管生成,淋巴管的生成弥补了淋巴管收缩能力的降低并使得区域淋巴流量增加。而在全身性炎症的急性状态下,如脓毒症,淋巴管的生成可能不足以克服炎症引起的淋巴松弛的全身效应。因此,炎症引起的淋巴引流减少可能是脓毒症期间液体失衡的主要因素,并被一些作者视为治疗靶点。
此外,淋巴系统的改变在器官损伤的发展中也起着关键作用,并可能阻碍愈合机制。在肺部,淋巴循环的作用在肺移植中得到证实,肺移植时中断了淋巴管。肺移植受者表现出持续性水肿,然而,对淋巴管和淋巴管新生在原发性移植物功能障碍中能起多大作用,目前尚存争议。肺水肿的吸收对ARDS预后至关重要。通过跨膜泵,肺泡腔的液体转运到间质。在LPS诱导的急性肺损伤中,淋巴管新生标记物增加与改善液体清除和生存相关。淋巴在急性器官功能衰竭中的作用在急性肾损伤(AKI)的小鼠模型中得到证实,其中淋巴扩张改善了恢复,暗示淋巴管生成在防止AKI进展为慢性肾脏疾病中的重要作用。尽管在人类脓毒症中缺乏临床数据,但淋巴引流的作用在脓毒症的消退阶段显然非常重要,无论对器官功能障碍恢复还是常见液体过负荷的吸收都至关重要。
结论
间质并未被动旁观,而是在调节跨毛细血管液体流动中最终崭露头角,特别是其在抽取血管内液体的能力方面。在局部炎症和肿瘤环境中,间质对毛细血管过滤调控中的重要作用证据越来越多,但脓毒症时,间质的参与证据仍稀缺。脓毒症时,关于其确切作用仍然存问题多多:压力变化的幅度是多少?是否有器官特异性反应?皮下成纤维细胞和脂肪细胞在细胞因子分泌中的各自作用是什么?细胞外基质(ECM)损伤在水肿的形成和消退中的作用是什么?在当前个性化的医疗时代,研究这个鲜为人知的领域可能会带来有价值的见解,增强我们对病理生理学的理解,并为探索新的生物标志物和治疗方法开辟独特的视角。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#微循环# #脓毒症休克# #流体动力学#
84