万分之一的变数,可能是一个令人激动不已的生命奇迹,也可能是一个令人迷惑不已的医学难题。比如,反复水肿40余年,出现的症状时而像皮肤过敏,时而又像阑尾炎,时而喉头水肿发作......先后辗转多家医院,相继被诊断为过敏、阑尾炎等进行对症治疗,却始终不见效果!
到底是什么样的疾病令人如此迷惑?40多年的谜团背后真相究竟为何?
反复水肿40余年,曾被误诊为阑尾炎
患者基本情况:
女性,57岁。
主诉:
身体不同部位反复水肿发作40余年。
现病史:
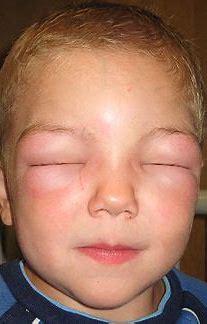
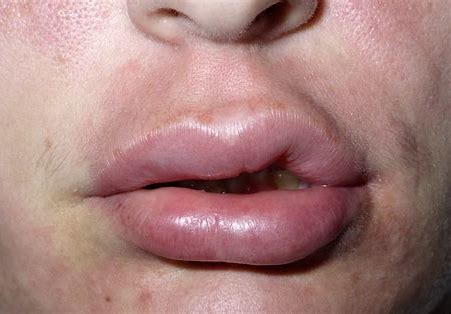
患者主诉青春期时,每月发生水肿1-2次,发作迅速,部位表现为游走性,时而手部、足部,时而面颊部、喉部,喉部不适表现为勒紧束缚窒息感,出现胃肠道不适时表现为腹痛、腹胀。面部、手足部水肿能耐受,可在3-4天内自行消退,曾因喉部、胃肠道不适前往医院就诊,多以“消炎止痛”处理,具体用药不详。
曾服用“氯雷他定”,症状无明显缓解后便未再使用。每次发作前无预兆,但多数发作前多伴劳累或烦躁。
曾于2003年至2004年期间,因腹痛、腹胀于云南某医院急诊行“开腹探查”,最终行“阑尾切除术”。其后,患者仍有上诉症状出现。
既往史:
过敏性鼻炎、胆囊切除术后。
家族史:
父母、弟弟、女儿无类似症状出现。
实验室检查
过敏原检测:
总IgE>100U/mL;
粉户尘螨特异性IgE抗体 弱阳性(+);
猫、狗皮屑特异性IgE抗体 阳性(++及以上);
霉菌组合1(点霉菌/烟曲霉) 弱阳性(+)。
注:此次就诊因为水肿而非皮肤发痒、风团等。
当您读到这里,结合患者的病史及部分实验室检查,您认为这位患者可能所患何种疾病呢?
补体C4测定:
0.043 g/L(参考区间0.1-0.4 g/L)
C1酯酶抑制剂浓度测定:
0.06g/L(参考区间 0.21~0.39g/L)
至此,实验室检查结果更加详细,您是否已知患者可能所患何种疾病?
答案揭晓
结合患者的临床表现及实验室检查,该患者在四川大学华西医院被确诊为遗传性血管性水肿(hereditary angioedema, HAE),但由于患者长期生活在昆明,因此于昆明医科大学第一附属医院就诊并接受相应的治疗。
病例讨论
HAE是一种罕见的常染色体显性遗传病。临床上以反复发作、难以预测的皮肤和黏膜下水肿为特征。水肿可累及身体任何部位, 以四肢、面部、生殖器、呼吸道和胃肠道黏膜较为常见。其中,最致命的是上呼吸道黏膜水肿(upper airway angioedema, UAE), 可因喉水肿迅速进展导致呼吸困难或窒息, 如抢救不及时可窒息死亡[1]。HAE的发病机制主要是由于C1酯酶抑制物浓度和(或)功能出现异常引起的缓激肽过量释放,血管通透性增加引起的水肿(图1 )[2]。
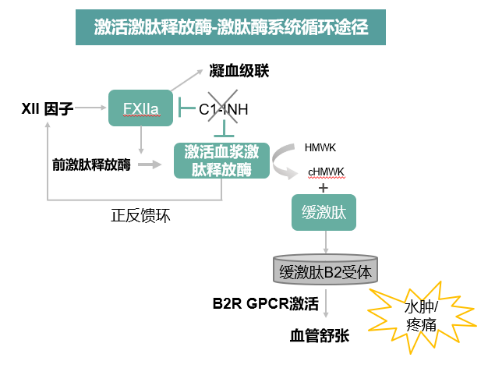
图1. 激活激肽释放酶-激肽酶系统循环途径
作为一种罕见病,HAE在医生和大众中的知晓率低,经常被漏诊误诊误治。北京协和医院研究表明,患者从发病到明确诊断,约需13年[3]。若能及早确诊,可显著改善患者的生存质量。针对HAE的诊断,《国际WAO/EAACI遗传性血管性水肿管理指南》(2021版)指出[4]:
诊断:
当患者出现反复发作的皮肤肿胀(四肢、面部和生殖器)、胃肠道发作(腹痛)和/或喉水肿时,应疑似为HAE。
当患者报告发生以下任何或所有情况时,进一步疑似HAE:
(1)阳性家族史(尽管约25%的患者可能没有);
该患者父母、弟弟、女儿无类似症状出现,暂未观察到阳性家族史。
(2)在儿童/青少年时期出现症状;
该患者青春期时开始水肿发作。
(3)反复发作的腹痛;
该患者出现胃肠道发作时表现为腹痛、腹胀。
(4)上呼吸道水肿;
该患者主诉喉部发作时表现为勒紧束缚窒息感。
(5)抗组胺药、糖皮质激素、奥马珠单抗和肾上腺素治疗无效;
该患者曾服用“氯雷他定”,症状无明显缓解。
(6)肿胀前出现前驱症状或体征。
(7)没有风团疹。
疑似HAE应立即进行实验室检查(C4、C1酯酶抑制物),以支持HAE的诊断。该患者测得补体C4和C1酯酶抑制物浓度均显著下降。基因检测并非确诊必需流程,但应患者本人要求,该患者进行了基因检测,结果显示:SERPING1基因存在杂合突变。SERPING1基因位于第11号染色体上,是合成C1酯酶抑制物的基因,该基因发生致病突变将影响C1酯酶抑制物的合成或功能,从而导致HAE的发病。
参考文献
[1] 支玉香, 安利新, 赖荷, 等. 遗传性血管性水肿的诊断和治疗专家共识[J]. 中华临床免疫和变态反应杂志, 2019,13(01):1-4.
[2] Nussberger, J et al. “Plasma bradykinin in angio-oedema.” Lancet (London, England) vol. 351,9117 (1998): 1693-7. doi:10.1016/S0140-6736(97)09137-X.
[3] Xu Y Y , Jiang Y , Zhi Y X , et al. Clinical features of hereditary angioedema in Chinese patients: New findings and differences from other populations[J]. European Journal of Dermatology, 2013, 23(4):500-504.
[4] Maurer M, Magerl M, Betschel S, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022;77(7):1961-1990. doi:10.1111/all.15214.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言