CDE发布全新指导原则,近400个生物类似药面临变局?
2020-03-20 铁皮憨憨 bioSeedin柏思荟

3月17日,国家药品监督管理局药品审评中心(CDE)发布公开征求《利妥昔单抗注射液生物类似药临床指导原则(征求意见稿)》意见的通知。时隔三年,CDE再次发布单品种研发制定审评要点。上一个单品种药物为贝

美罗华(Rituxan)是罗氏旗下基因泰克( Genentech )公司开发的一种CD20单克隆抗体,能够与B淋巴细胞上的CD20抗原特异性结合,能够引起B细胞的溶解,杀死癌细胞。
自1997年首次被美国FDA批准上市以来,美罗华已在全球范围内获批治疗非霍奇金淋巴瘤、类风湿性关节炎、慢性淋巴细胞白血病、滤泡性淋巴瘤、寻常型天疱疮、华氏巨球蛋白血症等多种适应症。2019年销售额达到66.6亿美元。
2000年,利妥昔单抗在中国上市,商品名为美罗华®,获批非霍奇性淋巴瘤、慢性淋巴细胞白血病等适应症。2017年,美罗华纳入国家医保目录,医保的支付价格为2418元(100mg/10ml/瓶)和8289.87元(500mg/50ml/瓶)。
随着专利在各国逐渐到期,世界各国仿制药企业都跃跃欲试,如诺华山德士、Celltrion和梯瓦等药企都想从利妥昔单抗上分一杯羹。
2019年2月25日,由复宏汉霖研发的利妥昔单抗注射液( HLX-01 )上市申请,该产品成为我国首个获批的生物类似药。据不完全统计,布局利妥昔单抗生物类似药的企业超过几十家,其中不乏正大天晴、华兰生物、海正药业、丽珠单抗、信达生物等知名企业,竞争非常激烈。
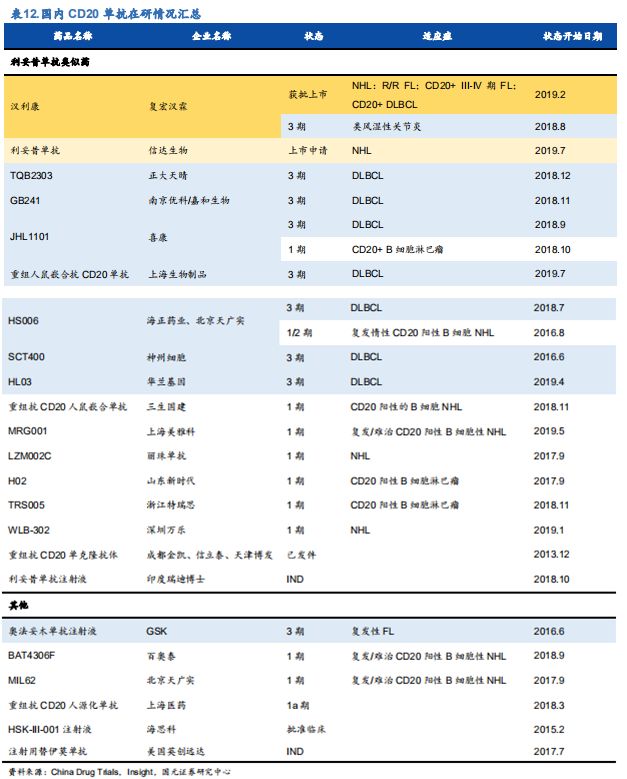
图片来源:国元证券《抱变革·制胜创新之三:
顾盼大分子市场——生物类似药篇》
经过对仿制药、创新药和生物类似药的研发过程的梳理和比较,不难发现和化学仿制药相比,生物类似药的研发技术壁垒较高,需要在证明药学相似的基础上证明药理学、临床药理学和临床相似。
生物类似药的平均开发年限为 5-9年,研发经费为4200万美金到10亿美金不等,研发年限和费用均高于化学仿制药。

图片来源:2019生物药开发者大会
《生物类似药临床研发与评价》-时岚
复宏汉霖刘世高博士也曾有过这样的一个比喻,“如果研发化学仿制药是‘造自行车’,那么研发生物类似药就相当于‘造飞机’。”
事实上,2009年,复宏汉霖启动利妥昔单抗生物类似药项目的时候,国内并没有相关的标准与指导原则——研发人员只能参考欧盟和美国的生物类似药相关法规,逐项摸索;直到2015年,原国家食品药品监管总局发布了《生物类似药研发与评价技术指导原则(试行)》,对生物类似药的申报程序、注册类别和申报资料等相关注册要求进行了规范,为企业研发提供了基本指导。

随着技术水平、政策监管及医保对价格的追求,为国内生物类似药带来了高速发展的契机。2017年10月,复宏汉霖向原国家食品药品监管总局递交新药上市申请并获受理,利妥昔单抗注射液成为国内第一个注册申请获得受理的单抗生物类似药。2019年2月正式获批,中间经历过多次临床研究策略及临床试验设计问题的讨论及数据的补充上交。
汉利康可以说伴随着国内生物类似药技术审评标准共同成长。
生物类似药与创新生物制品和化学仿制药的临床研究目的有所不同,生物类似药的目的是证明其与参照药在临床疗效和安全性方面的相似性,对应的临床试验设计围绕I期的PK/PD比对试验和III期的临床疗效和安全性比对试验来展开,其主要的评价指标是PK、PD终点、有效性终点与参照药的相似性。
化学仿制药的临床评价模式相对简单,进行生物等效性(BE)试验证明仿制药和参比制剂具有生物等效性即可,评价终点为PK终点生物等效。而创新生物制药最为复杂,其需要经过I-III期系统的临床试验来证明新药的安全性和有效性,评价指标为临床获益的有效性终点。
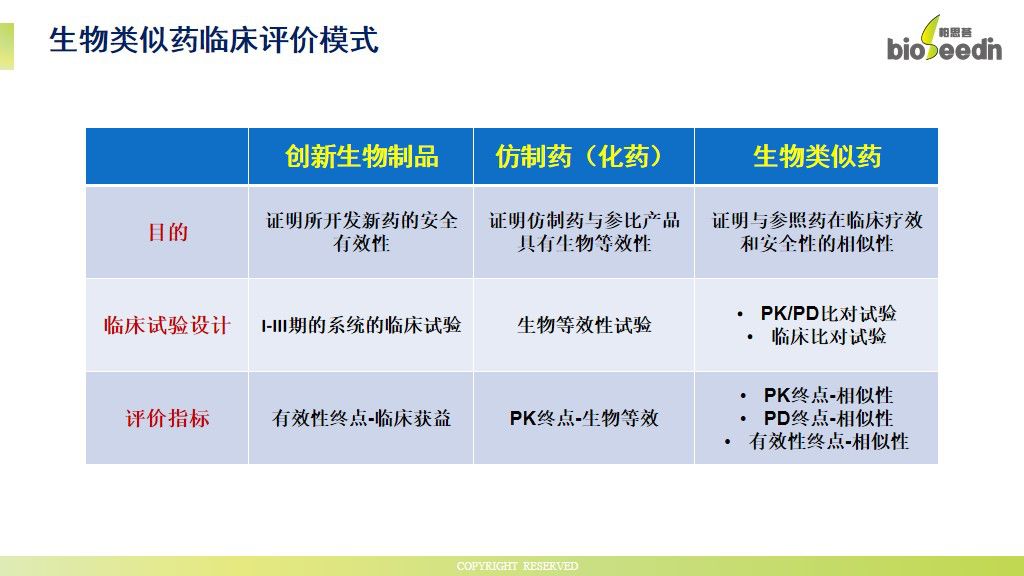
图片来源:2019生物药开发者大会
《生物类似药临床研发与评价》-时岚
以PK对比研究为例,阐述其试验设计、研究人群、参照药来源、剂量及给药途径、终点指标与界限、样本量等指导建议,并针对利妥昔单抗的特点推荐主要研究终点指标。
FDA在其生物类似药指导原则中静脉给药倾向于选择AUC(0-inf)作为主要研究终点。目前认为AUC0-Tau是通过实际测量值计算获得的,考虑到生物类似药的药代动力学特性和实际研究过程中取血点设计的相关性,推荐AUC0-Tau作为主要研究终点指标。AUC(0-inf)和Cmax作为次要研究终点重点进行比较分析。等效性界值根据常规建议设定为80%-125%。
1.生物类似药的研发应遵照比对、逐步递进、一致性和相似性评价的4大原则。
2.生物类似药的临床试验研究主要包括I期的药代动力学和/或药效比对试验(PK/PD)以及III期的临床疗效和安全性比对试验。
3.生物类似药和参照药的动物实验证明免疫原性相似、PK/PD、临床疗效和安全性相似都不等于两者的免疫原性相似。当前,生物类似药I期和III期临床研究中都需要进行免疫原性研究。
4.如果生物类似药和参照药的研究数据表明,在已经研究的适应证上与参照药达到可互换性要求并提供足够的科学证据,申请者可以申请参照药已批准的其他适应证也在生物类似药上得到批准。
5.可互换性是指生物类似药与参照药高度相似,以至于患者可以安全地从对照品转换到生物类似药,反之亦然(转换),并预期有相同的体验且安全性或疗效不发生改变。生物类似药的批准不是证明或者批准类似药和参照药可以互换,要想证明生物类似药与参照药具有“可互换性”,申办者一般需要开展药物互换的临床研究。
业内专家表示,从2015年国家出台了《生物类似药研发与评价技术指导原则(试行)》,希望人民可以用上更高质量的药物。尤其是像GPO、4+7集采政策实施以来,医保对药企的要求越来越高,越来越多质量好、价格合适的药纳入集采范围。
从国际上来看,2017年中国加入了ICH,中国对药品的管理越来越规范、成熟,让国内药企的产品在全球也有了一定的竞争力。指导原则的提出,也进一步规范了利妥昔单抗注射液生物类似药在临床试验的标准,对后续开发此类药物的企业有一定的指导意义,可以少走很多弯路。同样对其余生物类似药的临床试验有一定的启发性意义。
在未来,国内的制药企业既需要做好创新,与国外企业进行竞争;更需要开发适合国情的药物,造福国人。

图片来源:国元证券《抱变革·制胜创新之三:顾盼大分子市场——生物类似药篇》
参考内容
1. 国元证券《抱变革·制胜创新之三:顾盼大分子市场——生物类似药篇》
2. 时岚:2019生物医药开发者大会的演讲 《生物类似药临床研发与评价》
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#生物类似药#
63