
CMLS 天津医科大学杨洁教授团队揭示ELK1/SND1/SRF是促进VSMC表型转化和新生内膜增生的关键信号通路
2024-02-07 论道心血管 论道心血管 发表于陕西省

该研究首次揭示了SND1蛋白在VSMC表型转化及血管内膜增生中的作用及调控机制,为血管狭窄或再狭窄提供潜在预防和治疗靶点。
血管狭窄是心血管疾病最重要的病理过程之一,再狭窄是制约介入或搭桥治疗效果的主要障碍。血管平滑肌细胞(Vascular Smooth Muscle Cell,VSMC)是血管壁的主要成分,表现出高度的可塑性和动态变化能力,在外界损伤刺激下可由静息、收缩状态转化为增殖、合成状态,从血管中层迁移至内膜,并释放大量胶原蛋白等细胞外基质,加剧内膜增生,导致血管狭窄,在血管疾病中发挥关键作用。因此,研究VSMC表型转化的分子机制和关键调控因子对于精确预防血管狭窄或再狭窄至关重要。
Staphylococcal nuclease domain-containing protein 1 (SND1),又称为p100或Tudor staphylococcal nuclease (Tudor-SN),是一个保守的多功能蛋白,参与细胞增殖和分化的调控。它在有增殖能力的细胞和组织中高表达,在终末分化或静止期的细胞中表达水平显著降低。然而其在VSMC表型转化和血管疾病中的作用机制尚未阐明,需要进一步深入研究。
2024年1月27日,天津医科大学杨洁教授团队在Cell Mol Life Sci上发表了题为“Vascular injury activates the ELK1/SND1/SRF pathway to promote vascular smooth muscle cell proliferative phenotype and neointimal hyperplasia”的研究论文,该研究首次揭示了SND1蛋白在VSMC表型转化及血管内膜增生中的作用及调控机制,为血管狭窄或再狭窄提供潜在预防和治疗靶点。

首先,为了检测VSMC表型转换过程中SND1表达变化,研究人员建立了大鼠颈动脉和小鼠股动脉导丝损伤模型。免疫组化结果显示,与假手术组相比,导丝损伤组新生内膜中SND1表达明显上调。Western blot进一步显示,损伤动脉组织中SND1的表达增加,同时mRNA水平也上调。损伤动脉中增殖细胞核抗原PCNA水平的升高以及收缩表型标志蛋白ACTA2、CNN1和MYH11水平的降低表明了VSMC从收缩状态向增殖表型的转变。
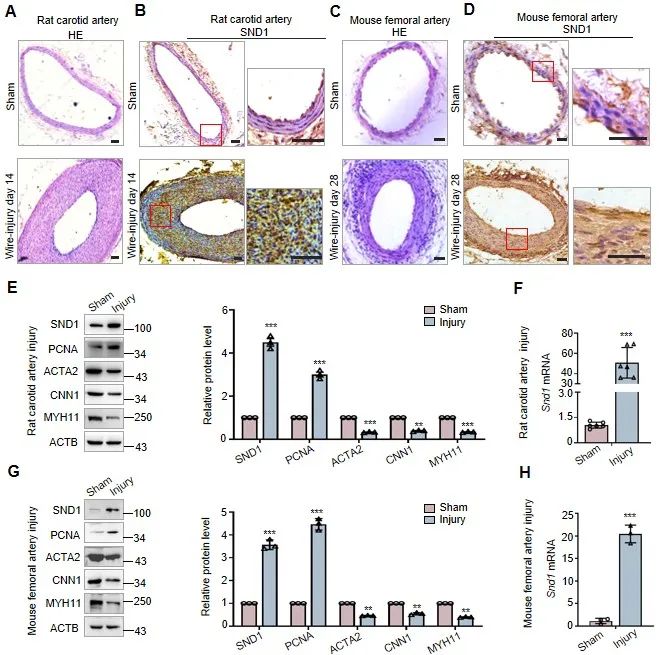
图1. 血管损伤后新生内膜中SND1表达增加
随后,研究人员利用主动脉离体培养模型及PDGF诱导模型,体外诱导VSMC表型转化。与新鲜提取的主动脉组织(Fresh)相比,离体培养后的主动脉组织(Cultured)中增殖标志PCNA和SND1水平均上调,同时收缩标志蛋白ACTA2、CNN1和MYH11的蛋白水平下调。同样,在PDGF刺激下,VSMC从收缩状态向增殖表型转变,SND1的蛋白和mRNA水平均增加。
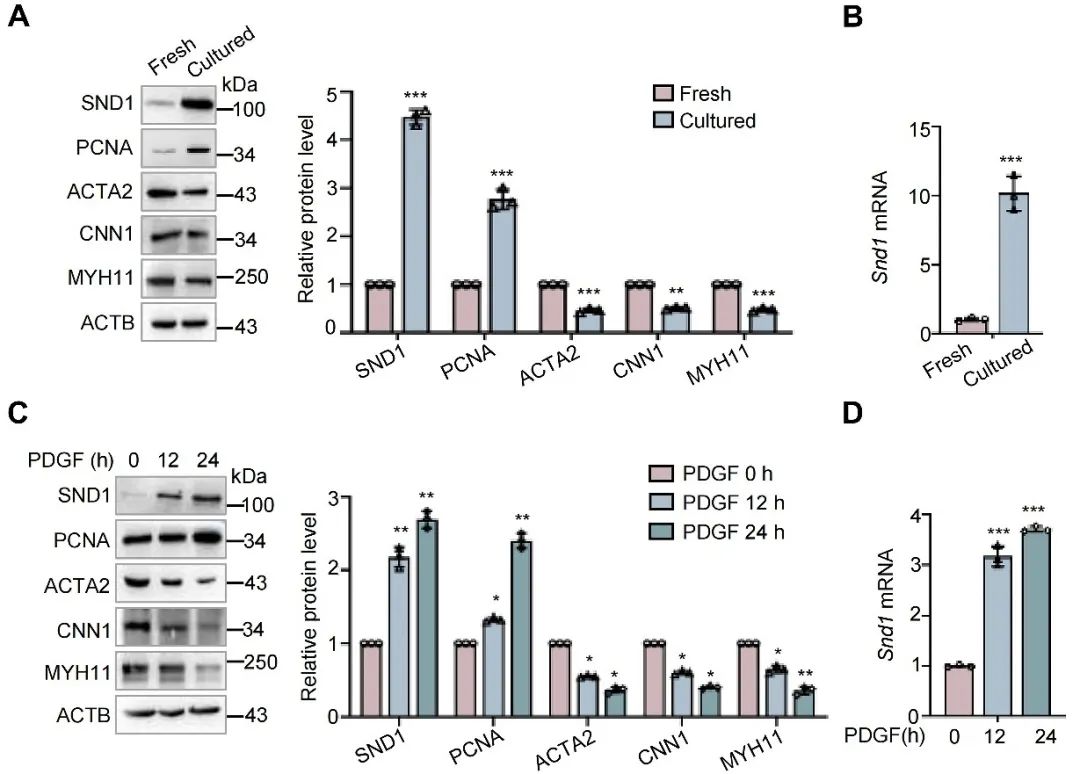
图2. 体外实验证明增殖型VSMC中SND1表达增加
为了评估SND1在VSMC表型转化及内膜增生中的潜在作用,研究人员构建了VSMC特异性SND1敲除小鼠(Snd1-F/F Tagln-Cre),然后构建小鼠股动脉导丝损伤模型。假手术组Snd1-F/F Tagln-Cre和Snd1-F/F小鼠的股动脉血管形态无明显差异。导丝损伤组Snd1-F/F小鼠新生内膜增生明显,而Snd1-F/F Tagln-Cre小鼠内膜增生被抑制。此外,与Snd1-F/F小鼠相比,导丝损伤Snd1-F/F Tagln-Cre小鼠动脉样本中增殖标志蛋白Ki67的染色减弱。以上结果表明,敲除SND1显著抑制了导丝损伤诱导的VSMC表型转化和内膜增生。
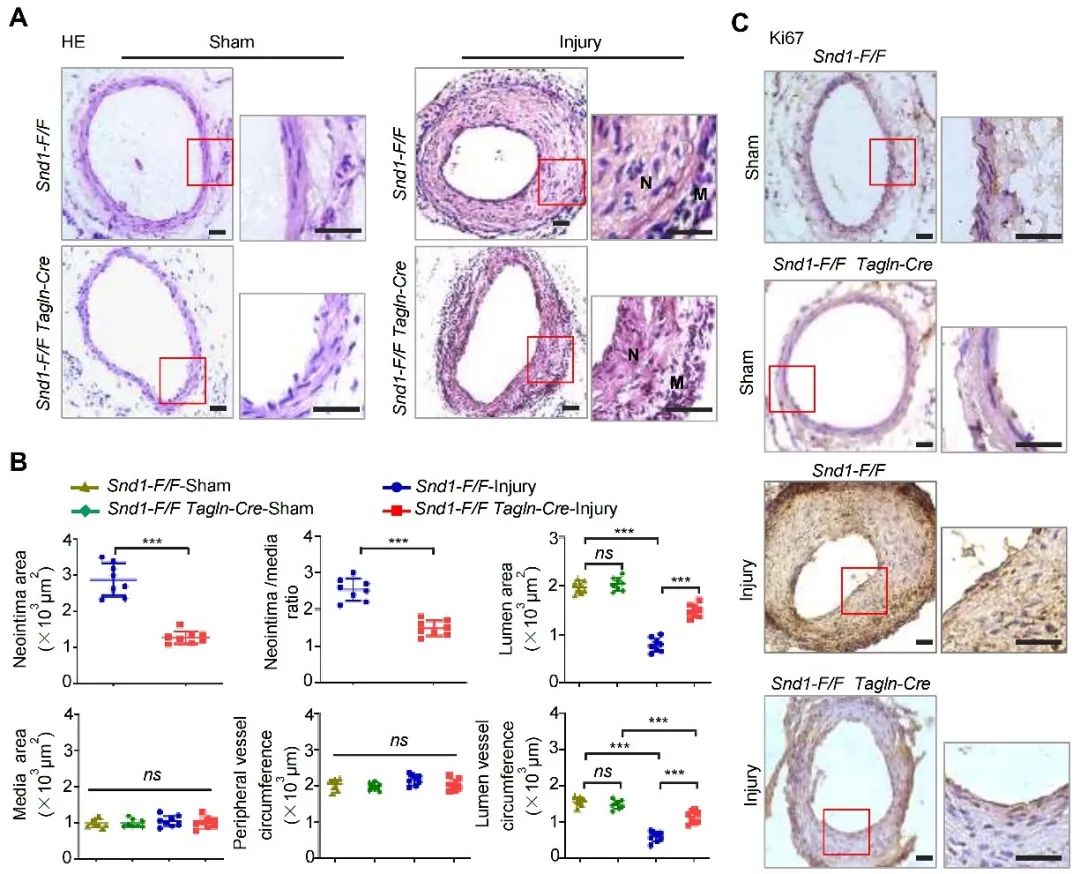
图 3. SND1敲除显著抑制导丝损伤诱导的VSMC表型转化和内膜增生
研究人员进一步利用原代VSMC检测SND1对增殖、迁移和收缩能力的影响。EdU掺入实验和CCK-8实验证实,SND1的敲除降低了原代VSMCs的增殖能力。划痕实验显示,敲除SND1降低了VSMCs的迁移能力。为了排除增殖对迁移的影响,研究人员利用活细胞成像来跟踪单个细胞的迁移。结果显示,Snd1-F/F Tagln-Cre VSMCs与Snd1-F/F VSMCs相比迁移能力降低。胶原收缩试验结果显示,敲除SND1可以提高VSMCs的收缩能力。
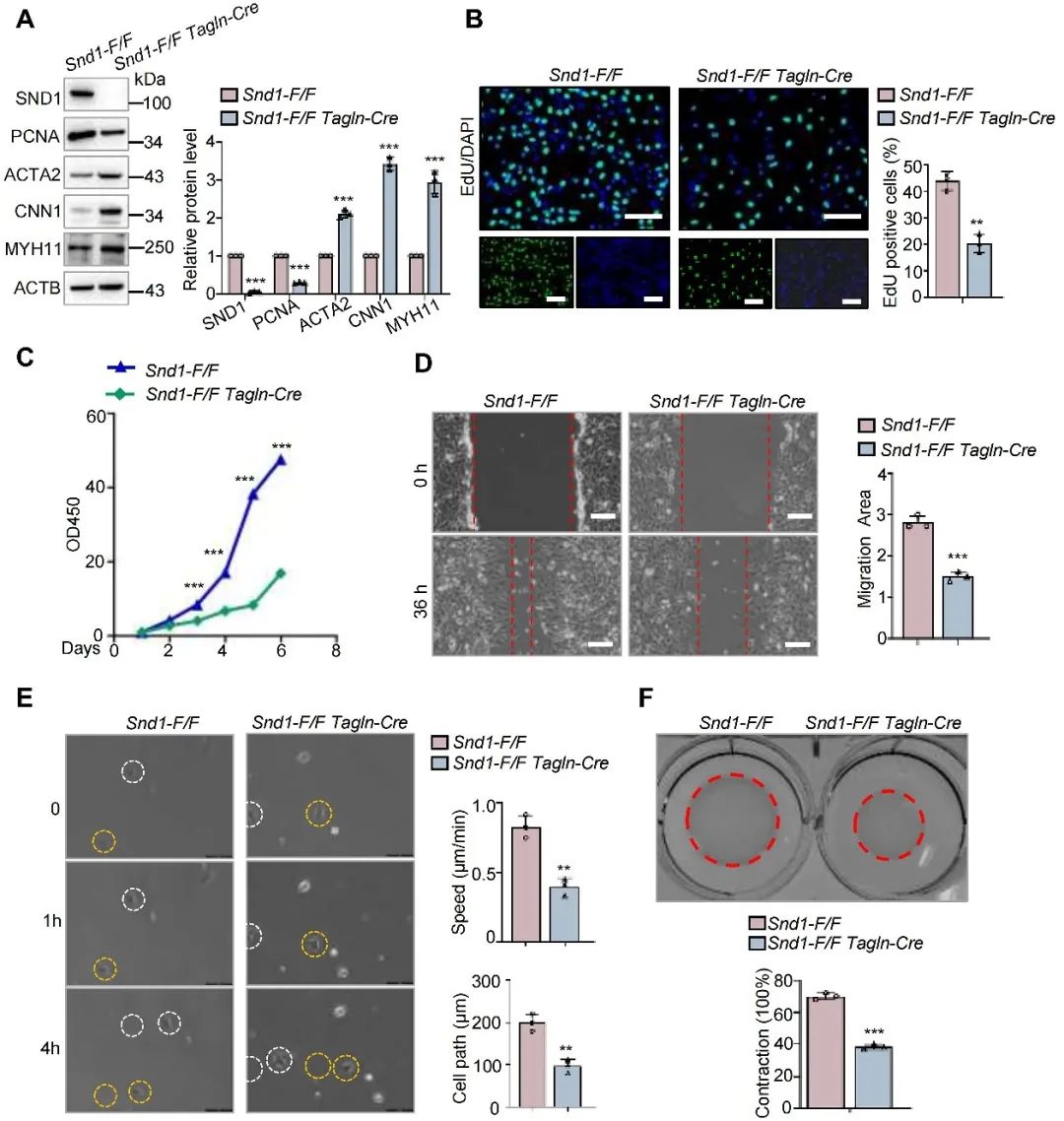
图4. 体外实验证明敲除SND1可抑制VSMC增殖表型
为了探索SND1促进VSMC增殖表型的分子机制,亲和纯化和质谱技术用来鉴定SND1的结合蛋白,结果显示SND1蛋白可以富集到调控细胞增殖和分化的重要转录因子SRF。Co-IP进一步验证了SND1和SRF之间的相互作用,且在PDGF处理或血管损伤后,SND1和SRF之间的结合增强。GST-pull down实验表明,SND1的SN结构域与SRF的MADS box结构域相互结合。
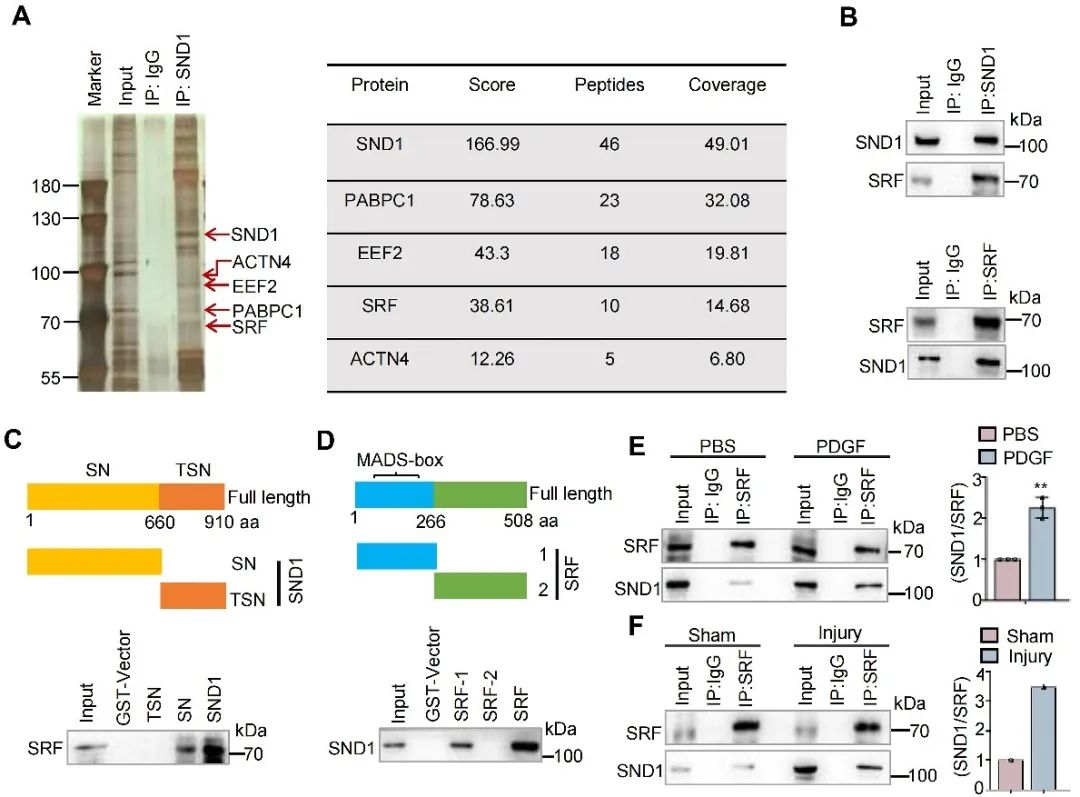
图5. SND1与SRF相互结合
凝胶电泳迁移实验(EMSA)显示,SND1依赖SRF与下游靶基因的CArG模序结合。染色质免疫共沉淀实验(ChIP)进一步证实,SND1影响SRF与下游增殖、迁移相关靶基因启动子区CArG模序的结合。双荧光报告基因系统检测SRF靶基因的转录活性,结果显示敲除SND1显著降低了SRF下游靶基因的转录活性,WB及PCR显示这些基因的mRNA和蛋白水平降低。
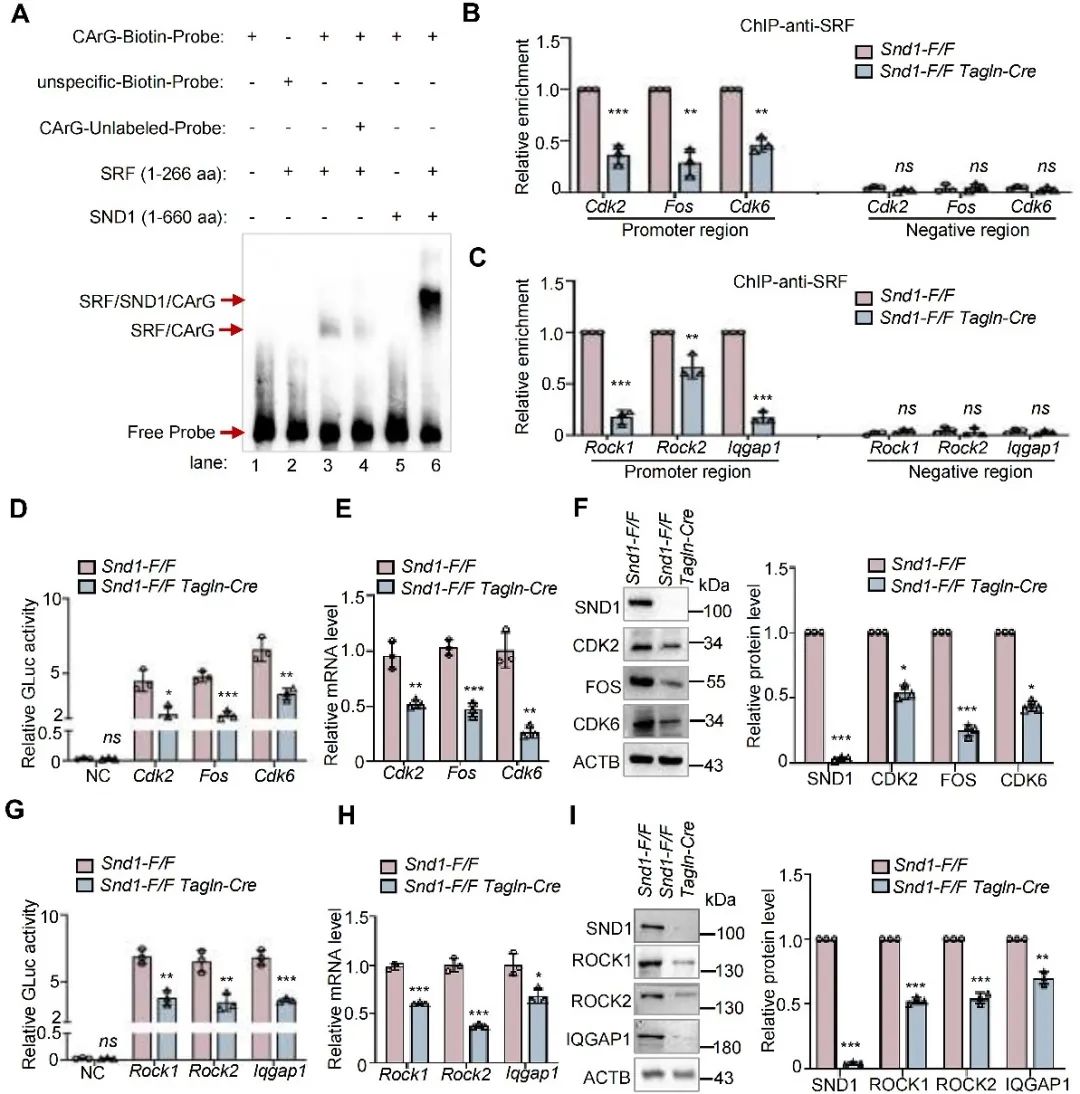
图6. SND1与SRF-CArG复合物结合促进VSMC增殖和迁移相关基因转录
研究人员进一步通过Co-IP实验证明了SND1可以与组蛋白赖氨酸乙酰转移酶lysine acetyltransferase 2B (KAT2B)结合,并影响SRF与KAT2B间的相互作用。组蛋白乙酰化ChIP实验显示,SND1的缺失导致乙酰化组蛋白H3K27ac和H3K9ac在增殖基因和迁移基因启动子上的富集显著降低。以上结果说明,SND1作为SRF的转录共激活因子,通过募集KAT2B,促进组蛋白乙酰化及下游基因转录活性。
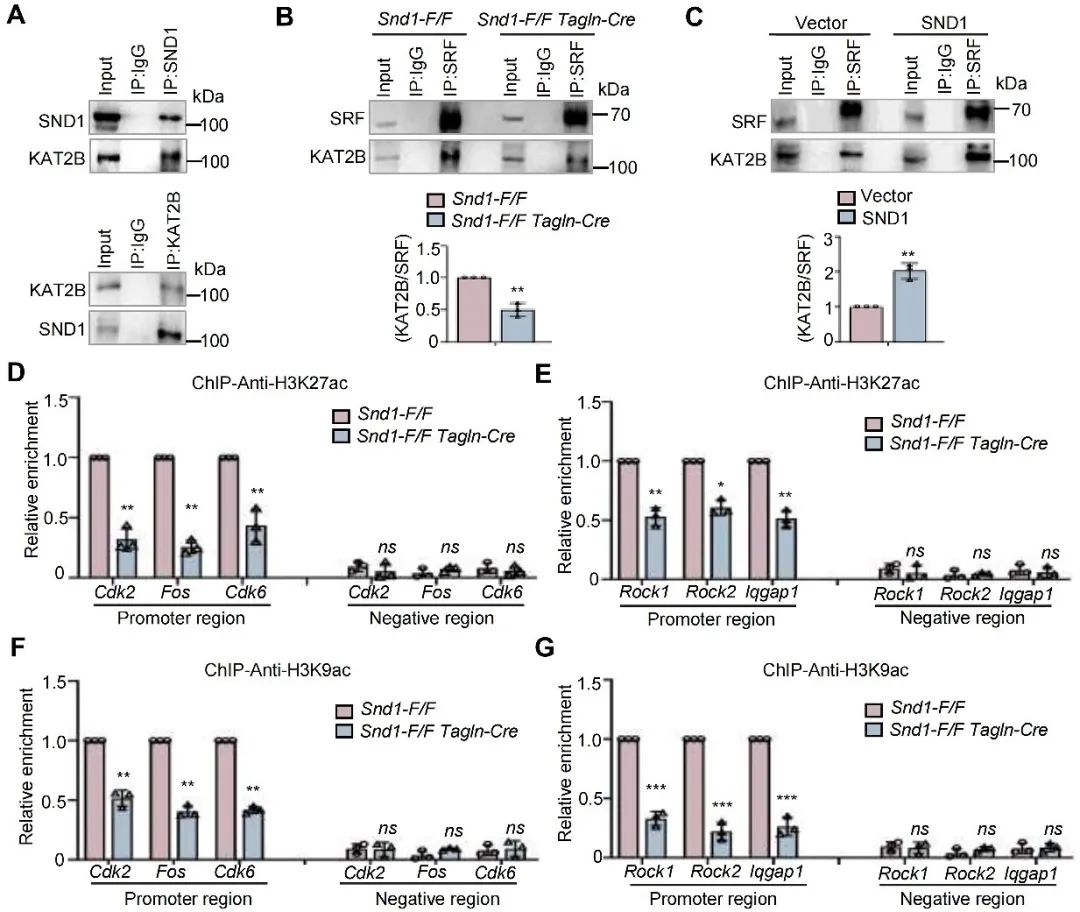
图7. SND1通过招募KAT2B作为SRF的共激活因子
利用生物信息预测的方式,研究人员发现ELK1是SND1潜在转录因子,并且SND1启动子区含有ELK1结合模序(-CCGGAAGT-)。EMSA及ChIP实验验证了ELK1与SND1启动子区特异模序的结合,并证实了在血管损伤或PDGF刺激下此结合的增强。双荧光素酶报告基因实验表明ELK1调控SND1的转录活性,WB和PCR结果显示ELK1对SND1表达水平的影响。这些发现支持了ELK1是SND1新的转录因子,调控SND1在PDGF刺激下的转录激活。
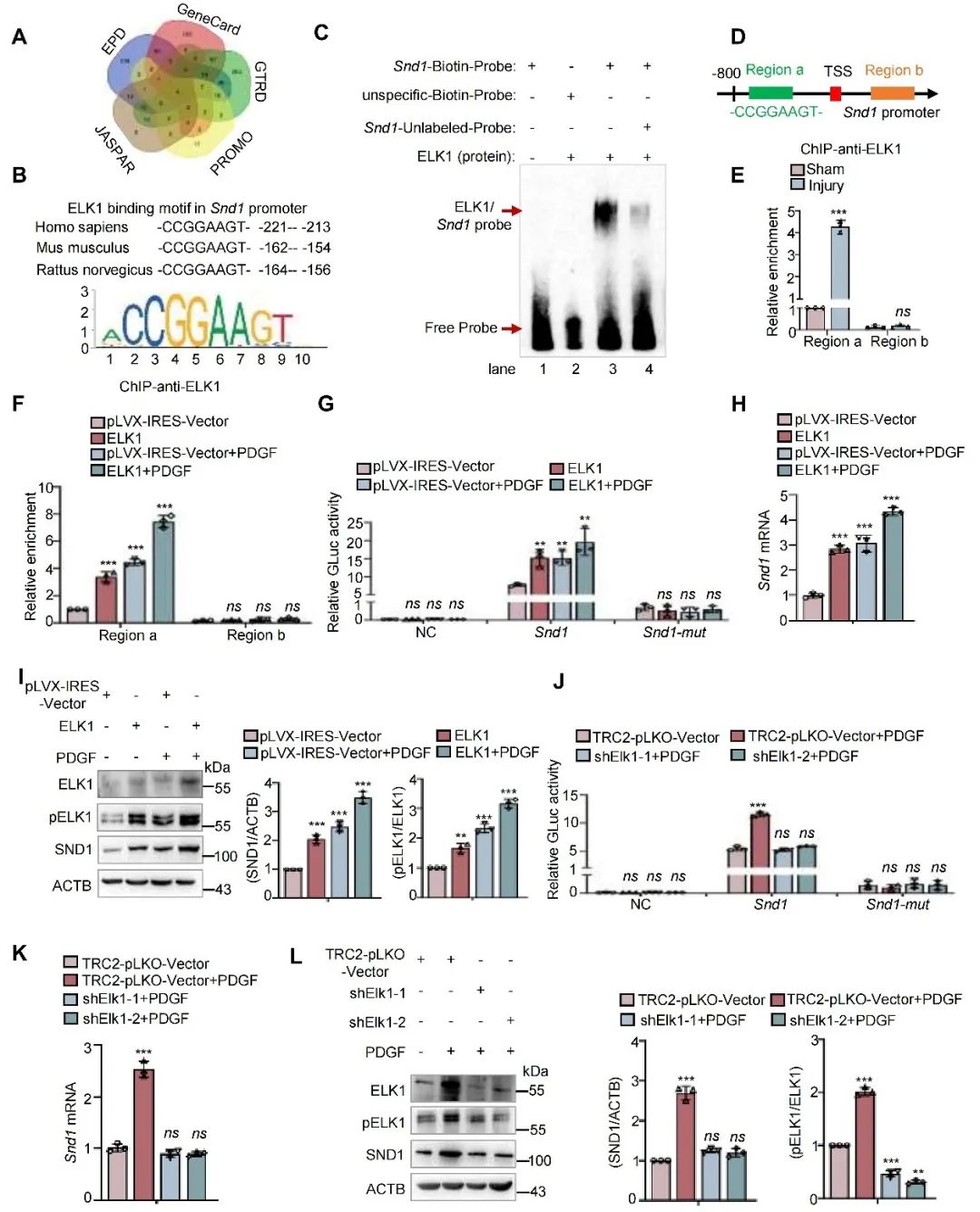
图8. ELK1促进VSMC增殖过程中SND1的转录激活
U0126是ERK/ELK1信号通路的有效抑制剂。研究人员构建小鼠股动脉导丝损伤模型的同时给予U0126治疗。结果显示,U0126抑制了损伤股动脉中ELK1的磷酸化和SND1的上调,并抑制了损伤诱导的新生内膜增生。进一步研究揭示U0126处理阻止了ELK1与SND1启动子区域的有效结合并降低了SND1的转录激活。这些结果进一步提示,U0126诱导SND1下调是抑制新生内膜增生和血管狭窄的潜在治疗策略。
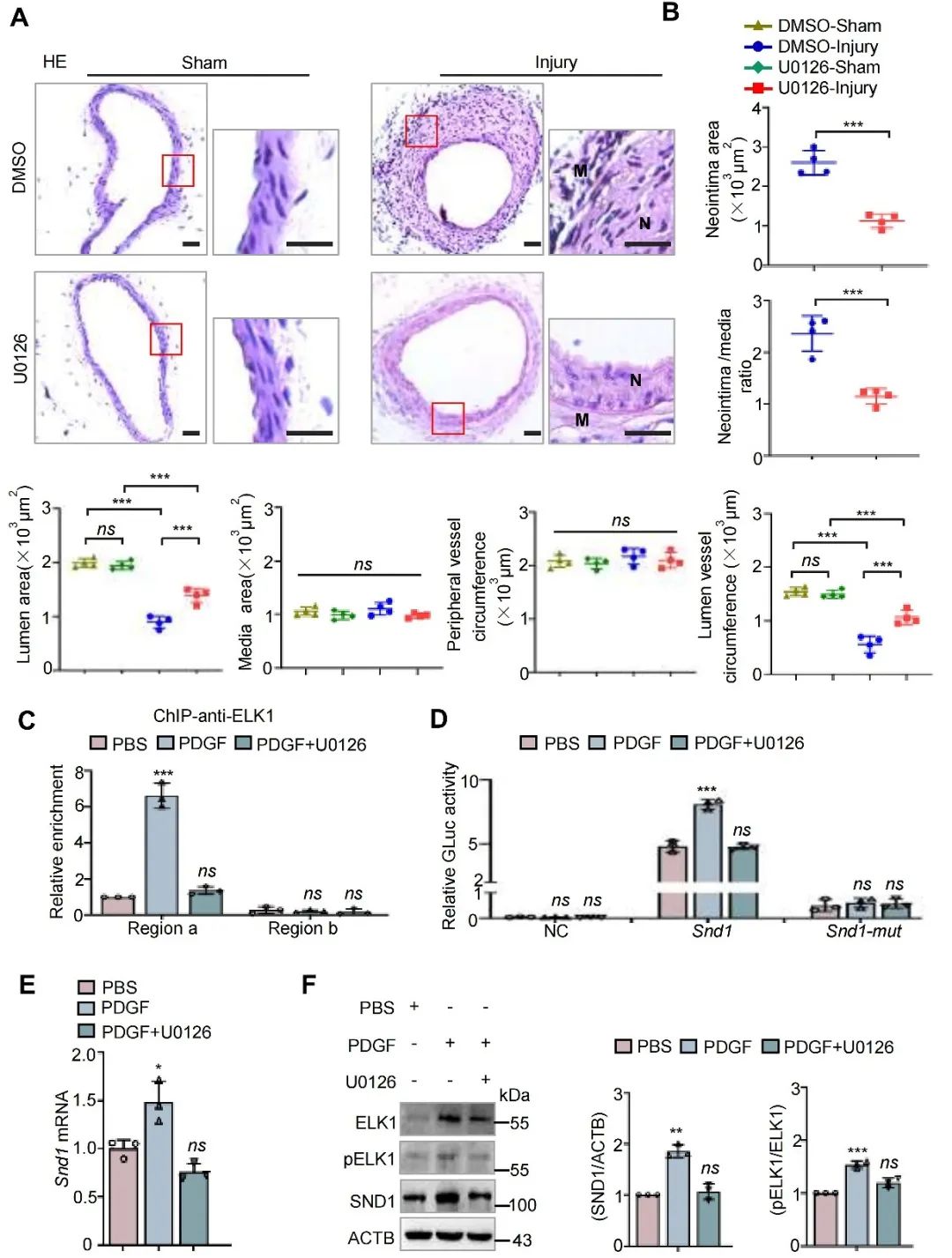
图9. U0126降低损伤诱导的SND1上调和新生内膜增生
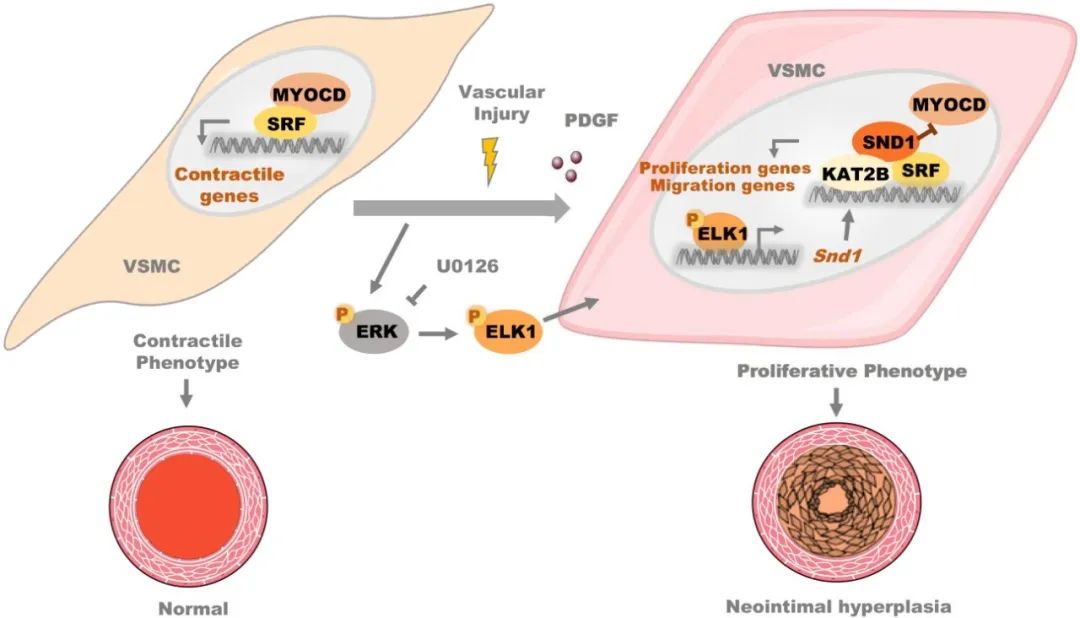
综上所述,本研究发现血管损伤时转录因子ELK1促进SND1的转录激活,上调的SND1作为SRF的共激活因子,募集KAT2B到下游基因启动子区域,促进组蛋白乙酰化,协助SRF识别特定CArG基序,增强增殖和迁移相关的基因转录激活,从而促进VSMC表型转化。ELK1/SND1/SRF是促进血管损伤中VSMC表型转化和新生内膜增生的新的信号通路。本研究为阻止内膜增生及血管狭窄提供了一个新的潜在治疗靶点。
天津医科大学杨洁教授及香港大学深圳医院魏民新教授是该论文的共同通讯作者。香港大学深圳医院苏超博士后、天津医科大学刘明霞博士以及姚旭阳博士后为该论文的共同第一作者。
原文链接:
https://link.springer.com/article/10.1007/s00018-023-05095-x
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#内膜增生# #血管狭窄# #SND1蛋白#
46