蛋白质稳态失衡(特别是蛋白质降解和清除减少)是阿尔茨海默病的一个明显特征。在神经退行性疾病研究中蛋白质稳态失衡对神经元回路功能障碍的影响是一个新兴的概念,对解释疾病造成认知能力下降的现象至关重要。近期《Molecular Neurodegeneration》发表了“Proteostasis failure exacerbates neuronal circuit dysfunction and sleep impairments in Alzheimer’s disease”的综述文章,论述了睡眠中断和蛋白质稳态失衡的双向关系对阿尔茨海默病进展的影响。
在文章中作者论述了阿尔茨海默病的蛋白质平衡功能障碍和tau蛋白病症破坏了调节睡眠-觉醒周期的神经元,并在行为上表现出慢波和快速眼动睡眠模式受损。而随后的睡眠不足和昼夜节律紊乱会通过损伤蛋白酶体、自噬、未折叠蛋白反应和淋巴清除,进而增加β-淀粉样蛋白和tau蛋白的传播。作者认为睡眠不足应作为一系列神经退行性疾病和混合神经病理病例的重要考虑因素,并建议将增强睡眠疗法作为促进健康蛋白质平衡的潜在干预措施进一步进行临床和临床前研究,为阿尔茨海默病和其他脑部疾病的精确单一和组合治疗提供信息。

(1)睡眠-蛋白质平衡双相关系的理论依据
老龄化的神经退行性疾病(NDDs)有一个共同的蛋白质病变机制,即有毒的、错误折叠的蛋白质积累,在整个大脑的播种增加,并聚集在细胞外和细胞内包涵体中。蛋白病的治疗必须克服三个挑战:首先,蛋白病的发生有长达数十年的前驱期,而诊断主要发生在晚期阶段;其次,由于晚期广泛的蛋白病变,尽管介入了相关治疗但神经变性仍然存在;最后,正常清除蛋白的内源性过程受损,限制了异常蛋白清除的有效性和持续性。
了解阿尔茨海默病中β-淀粉样蛋白(Aβ)和tau蛋白在疾病进展早期阶段的扩散机制是至关重要的。尽管Aβ和tau蛋白对神经元电生理有不同的影响进而损害了行为表型(包括认知和睡眠),但神经变性和认知能力下降都与tau的区域积累密切相关。目前阿尔茨海默病的神经网络功能障碍模型确定,神经元过度活跃是Aβ病理的结果,而tau蛋白被证明是抑制神经元活动的。与野生型对照相比,带有斑块的 APP/PS1 小鼠的第 2/3 层神经元表现出过度活跃;而尽管表达聚集人 tau (P301L) 的小鼠没有 Aβ 病理表型,但却表现出皮质神经活动水平的显著降低。对体外颞叶皮层 (EC) 切片共同使用Aβ 和 tau处理证明了可溶性 tau 对神经元活动的抑制作用(不依赖于神经纤维缠结),并且这种作用比 Aβ 诱导的神经元过度活跃更有优势。这些实验结果也得到了其他临床前模型证据的支持。Tau 病理变化导致年老(而非年轻)的 EC-Tau 小鼠出现空间记忆缺陷,其中内侧 EC 的兴奋性而非抑制性神经元显示更容易受 tau 病理学影响。有证据表明, Aβ 的存在促进了 tau 对小鼠神经元回路功能障碍的影响,而 Aβ 诱导的神经元的过度兴奋也依赖于 tau。这些影响如何在 AD 患者中呈现可能取决于 Aβ 和tau在不同区域的分布,是 NDDs 研究的一个新兴领域。
睡眠数量和质量的下降,尤其是慢波和快速眼动睡眠 (SWS;REM)与AD的病理发展有关。这些改变可能在很早期就开始发生,并会增加发生认知障碍和 AD 的风险。蛋白质稳态的许多方面表现出睡眠调节和节律性活动的变化。蛋白质降解和清除在 AD 中受损,并且与睡眠密切相关。特别是神经元活动周期的增加促进了 Aβ 和 tau 扩散,随着SWS的丧失会导致淋巴清除减少,睡眠和昼夜节律紊乱会导致未折叠蛋白反应(UPR)、泛素蛋白酶体系统(UPS)和自噬-溶酶体途径(ALP)的失调。蛋白质稳态的失调,包括未消化的自噬体和溶酶体的积累以及Aβ或tau等蛋白病,会导致受 AD 影响的脑区的神经变性,包括 EC、海马、下丘脑和蓝斑 (LC) 。这种神经变性反过来又会损害调节记忆和睡眠的神经回路。此外,Aβ 和 tau 分别影响睡眠状态和调节昼夜节律。总的来说,睡眠和蛋白质稳态之间的动态关系意味着一种机制的损害会加剧另一种机制,并共同加速 AD 进展。
总之,在 AD 中睡眠不足和蛋白质稳态障碍是相互影响的,涉及睡眠不佳和昼夜节律紊乱,这会损害与蛋白质清除有关的生物过程。然后,Aβ 和 tau 反馈通过控制睡眠-觉醒的神经元变性对睡眠-觉醒周期产生直接和间接影响。这个循环在整个 AD 进展中不断重复。鉴于近十年来的经验证据表明睡眠是蛋白质平衡的有效调节因子,因此睡眠-蛋白质平衡关系可能在阿尔茨海默病的早期阶段至关重要。而a β和tau的积累会造成睡眠相关神经元电生理特征受到损害,从而加剧了蛋白质病变。
(2)睡眠障碍是阿尔茨海默病的一个风险因素
与睡眠障碍和 NDDs风险增加有关的因素对于识别脆弱人群和解决睡眠不足问题至关重要。睡眠是AD的一个潜在可改变的风险因素。AD 的遗传风险会缩短睡眠时间,平均每晚少睡 1.87 小时。最近的一项荟萃分析得出结论,广泛的睡眠障碍(即质量差、失眠、睡眠不足/过度、睡眠呼吸暂停、白天过度嗜睡)导致 AD 的相对风险增加 1.55 倍,认知障碍提高 1.65 倍,初步判断为临床前 AD增加了3.78倍 。例如,50 - 60 岁且可能处于临床前 AD 阶段的个体睡眠不足(< 6 小时)会使痴呆风险增加 30% 。阻塞性睡眠呼吸暂停 (OSA) 是成人睡眠障碍的最常见原因,对 AD 构成显著较高的分组风险 (2.37倍),并且与淀粉样蛋白、tau 蛋白和神经退行性变性 (A/T/N) 的生物标志物对AD具有协同不利影响,其中 AD 蛋白病驱动的海马变性可能会加剧呼吸问题和夜间呼吸暂停。此外,轻度认知障碍 (MCI) 与整个睡眠阶段的睡眠重大改变相关,包括整夜觉醒和睡眠效率降低。总的来说,这些数据表明睡眠中断可以加速 AD 相关的神经变性,尤其在症状出现前阶段最为明显。
基因变化可能会增加睡眠相关的NDD和AD的风险。淋巴通路相关的水通道蛋白-4 (AQP4)的变异与小鼠模型中的AD病理和帕金森病(PD)的认知表现有关。AQP4 在脑脊髓液 (CSF) 中的含量较高,其失调可能引发神经变性(AD 和额颞叶痴呆 -FTD)。同样,与 ApoE3 相比,携带载脂蛋白 E4 (ApoE4) 等位基因并伴有睡眠呼吸暂停的男性的认知功能下降幅度更大。在基线没有痴呆的老年人中(平均年龄~82岁),更好的睡眠可降低 AD风险并减少 tau 病变/神经原纤维缠结的形成。这表明评估 AD 风险较高的 ApoE4 + 个体睡眠的重要性,并且通过睡眠引入干预措施可能会减少神经原纤维缠结负担。
目前大多数关于 AD 睡眠的研究(特别是那些研究 ApoE 等遗传变异作用的研究)都是在白种人中进行的,而对具有不同遗传风险特征的其他种族知之甚少。最近的一项工作发现,在老年黑人(平均年龄 ~ 70)中 ApoE4 基因型与 OSA有相互作用,与包括海马体积在内的 AD 生物标志物相关。而在白人参与者中未观察到这种相互作用。此外,与具有 E3 基因型的非裔美国人相比,至少具有一个 ApoE4 等位基因的非裔美国人的睡眠时间明显更短。尽管不分种族的年龄大于 50 岁的 ApoE4 + 参与者的睡眠中断较多,但在高加索人中未观察到这一现象。ApoE 基因型也可能与中国人群的 OSA 风险相关。尽管睡眠和蛋白质稳态的相互作用机制可能是一种全球现象,但未来的研究应该确定种族和民族在睡眠不足和蛋白质稳态失衡对 AD 进展贡献中所起的作用。
性别差异会影响个体患 NDD 的风险状况,在了解睡眠不足是如何影响NDD 进展时应考虑性别差异。AD 在女性中出现的频率更高。虽然男性更容易出现睡眠呼吸暂停,并且其他睡眠障碍在成年男性中比女性更常见,但绝经后女性的呼吸暂停增加。PD 和相关的突触核蛋白病、路易体痴呆 (DLB) 在男性中更常见,然而基于 REM 的障碍在女性生命后期往往比男性更容易发生。
与 AD 相关的确切睡眠障碍因个体和研究而异,但睡眠障碍在 AD 患者中很常见。其中日间过度嗜睡(EDS) 在 AD 患者中很常见,并且已被证明在轻度 DLB 和较轻程度的行为变异 FTD 患者中更为严重。这表明EDS 是老年人群尤其是痴呆症患者的共同特征。最近的证据已经建立了 Aβ 沉积和没有认知障碍的健康成人和老年人的 EDS之间的联系,使其成为 AD 潜在早期预测因子。值得注意的是,老年人的睡眠障碍与认知过程受损密切相关。简而言之,AD 与多种可能的夜间睡眠障碍有关,包括入睡时间延长、清醒时间和夜间觉醒时间增加、非快速眼动 (NREM) 2 期、SWS 和 REM 时间减少,以及 NREM 1 期增加; 尽管许多报告表明 NREM 阶段 3/SWS 减少和慢波振荡在 AD 中占主导地位。图 1 提供了健康夜间睡眠结构和分期的代表性示意图,它与记忆的相关性,以及与 AD 中发生的损伤的比较。

图1阿尔茨海默病的睡眠障碍示意图
(1)Aβ对睡眠的直接影响
AD 的两种主要病理蛋白都会对睡眠功能产生有害影响,Aβ 寡聚体以剂量依赖的方式干扰小鼠的睡眠/觉醒模式。最近,有学者通过将不同长度的Aβ寡聚体注射到斑马鱼中,描述了Aβ对其睡眠的直接影响。睡眠调节与 Aβ 寡聚体大小有关,其中短寡聚体增加下丘脑神经元活动并通过肾上腺素能和孕激素受体信号诱导清醒,长寡聚体减少神经元活动并通过朊蛋白途径诱导睡眠,而非常长的寡聚体则没有作用。相反,在小鼠中注射 Aβ 寡聚体会诱导睡眠片段化(减少 NREM 和 REM 时间并增加睡眠阶段转换),但这一现象在缺乏朊蛋白的小鼠品系中未观察到,说明 Aβ-睡眠调节的机制是复杂的。这些结果可能部分解释了与 AD 相关的广泛睡眠障碍,包括总睡眠时间减少和白天睡眠增加。此外,AD 中的睡眠和昼夜节律紊乱可能会引发这种睡眠的双向控制,其中清除受损Aβ会引起昼夜波动的改变。
(2)tau 蛋白与睡眠的关联性
最近的多项研究记录了 tau 蛋白与睡眠障碍和脑电图 (EEG) 异常的关系。在认知正常的老年人(平均年龄约 73-75 岁)中,NREM 睡眠期间的低频 EEG 信号(1-2 Hz;指示 SWS 中显著的 delta 波)与AD病理学呈反向关系;AV-1451 tau 正电子发射断层扫描 (PET) 信号和AD患者(轻度至中度)的睡眠-觉醒障碍已被证明与CSF磷酸化 tau 的水平相关。在小鼠和 健康30-60 岁成年人中睡眠剥夺会使大脑(小鼠)和脑脊液(人类增加 > 50%)tau和其它主要的 AD 生物标志物含量增加,其中 CSF pT181 和 pT217(但不是 pS202)含量更高,并伴随着这些表位的非磷酸化 tau 形式水平增加。
Winer 及其同事评估了 tau和 Aβ PET水平与老年人(平均年龄约 78 岁)睡眠的关联,比较了一周内客观(腕表活动记录仪)和主观(匹兹堡睡眠质量指数:PSQI) 测量数据。客观睡眠障碍与早期 Braak 相关阶段、EC 以及更广泛的内侧颞叶中更多的 tau PET 相关,但与皮质 Aβ PET 无关。然而,tau 和 Aβ 都与自我报告的睡眠障碍显著相关,表明更多的Aβ 个体报告的睡眠质量比他们实际的睡眠质量差,PSQI 整体和效率得分有显著变化,但睡眠持续时间没有变化。这种影响因执行功能的丧失而加剧。因此,主观上对睡眠质量的低估是否会影响未来客观睡眠障碍的进展是未来研究的一个有趣话题。
在患有OSA 的年轻个体中观察到tau的升高,与仅患有 OSA 的患者(年龄范围:35-65 岁)相比,同时患有 OSA 和 MCI 的患者的脑源性外泌体含有更高水平的总 tau、pT181 和 Aβ,这表明 NDD 外泌体介导的蛋白病传播对睡眠障碍有增效作用。总之,这些报告表明 tau 蛋白与睡眠的关联可能有助于在 AD 中观察到的蛋白病-睡眠双向关系。相反,其他研究表明,在健康的 20-40 岁人群中部分睡眠中断 5 天后总 tau 蛋白或磷酸化 tau(或 Aβ)没有变化。对健康志愿者(35-65 岁)进行过夜干预以防止 SWS 一晚,也没有显示 CSF tau 水平有任何显着变化,但 Aβ 升高。这些参与者6 天内在家睡眠的较差活动记录与 CSF tau 水平升高有相关性。值得注意的是,这两项实验都是在年轻和中年的健康参与者中进行的,随着年龄的增长和 NDD产生,内源性清除机制失效将导致睡眠不足后蛋白质积累增加。
脑电波(EEG)α波(7–12 Hz)是休息和安静觉醒的标志,并且已被证明与 AD 的tau病理相关。在主观认知能力下降、 MCI 和 60多岁AD 参与者中 EEG α波功率和同步性减弱与 CSF 总水平和磷酸化 tau 水平的增加相关,而 CSF 磷酸化 tau 与老年人(平均年龄70岁)的功率谱中α波具有较低的峰值频率有关。当按 Aβ (C-PIB) 和 tau PET (F-MK-6240) 阳性分层时,阳性组的峰值 α波频率从 ~ 9.5 Hz 减慢到 ~ 8 Hz。在认知正常的老年人(平均年龄约 75 岁)中(尤其是那些具有高 p-tau:Aβ 比率的人),CSF 总的和磷酸化 tau 蛋白与θ 波(以 4-8 Hz 分析)增加相关,但 δ、α 或 β 频率区间没有变化。静息激活的“默认模式网络”中的蛋白病和 tau 介导的神经元抑制可能是导致EEG减慢和 α波损伤。此外,α波效应可能因性别而分层:α 功率和总 tau 与 MCI(平均年龄 75岁)的男性而非女性参与者呈负相关,而在女性与男性健康老年人以及 MCI 和 AD(平均年龄 ~ 69-70)患者中报告了更多静息状态 α EEG 活动。这些结果表明 EEG α 具有作为非侵入性 AD 生物标志物的潜力,进一步调查研究 AD 中 α波活动受损是否会减弱安静清醒是非常有益的。
临床前模型支持tau蛋白与睡眠损失和脑电图改变之间的联系。在tau基因敲除小鼠中,δ功率、NREM和总睡眠时间减少,状态转换增加。在P301S(晚期)和rTg4510小鼠中,与睡眠改变相关的δ和θ脑电图功率降低,在FTD-tau小鼠模型中,清醒状态下的脑电图α功率降低,表明与tau相关的整体脑电图减慢。rTg4510皮质神经元电生理减慢,NREM睡眠上下状态受损。在 tau 蛋白病和 AD 模型中,睡眠剥夺增加了3xTg AD 小鼠的tau 纤维沉积以及tau 在 P301S 小鼠中扩散。最后,Tg4510 tau 蛋白病模型小鼠在视交叉上核 (SCN) 中表现出 tau 包涵体,并伴有 PER2 和 BMAL1 时钟蛋白的不规则表达。Tau缺陷的果蝇还表现出昼夜节律紊乱,活动模式异常,起搏器神经元的神经重塑受损,昼夜时钟蛋白增加。而表达 4R tau 的果蝇会出现昼夜节律失常和睡眠紊乱的行为。总之,AD 的 tau 病理和功能丧失因睡眠中断而加剧,并反过来通过昼夜节律失常和睡眠调节神经元群受损而损害睡眠。
(1)NDD血浆生物标志物和睡眠障碍
血浆生物标志物是早期检测与睡眠相关神经变性的潜在方法,但疾病诊断的金标准仍有待建立。在阿尔茨海默病中已取得部分进展,其中Aβ42、A β40、Tau-181、Tau-217和Tau-231以及炎症标志物胶质纤维酸性蛋白(GFAP)和神经退行性标志物神经丝轻链(NfL)都显示出应用前景。前三个追踪脑脊液中 Aβ 和 Tau-181 水平变化与睡眠障碍的关系表明睡眠障碍与神经变性有直接联系。呼吸暂停也与血浆 Aβ42/Aβ40 比率和磷酸化 tau 增加有关。神经变性的生物标志物可能代表睡眠调节的改变,包括 AD 患者血浆中食欲素 A 的增加和亨廷顿病 (HD) 患者血浆褪黑激素的减少。再加上前面提到的 CSF 中 AQP4 的增加,这些研究表明 NDD 中睡眠相关途径发生了功能障碍。
虽然大多数与睡眠功能障碍相关的生物标志物都集中在阿尔茨海默病相关的蛋白病上,但还有其他与睡眠相关的疾病生物标志物,包括血浆代谢组和脂质组。相对于年龄匹配的对照组,血浆 TNF-α 和 IL-10 在 REM 睡眠行为障碍 (RBD)(PD 前驱)中显著升高,此外IL-6/IL-10 和 IL-8/IL-10 水平降低。TAR DNA 结合蛋白 43 (TDP-43) 和 9 号染色体开放阅读框 72 (C9orf72) 聚集也出现在调节睡眠-觉醒周期的下丘脑和视交叉上核神经元中。TDP-43 和 PET 生物标志物的开发是一个活跃的研究领域,并且可能证明对肌萎缩侧索硬化症 (ALS)/FTD、边缘显性年龄相关 TDP-43 脑病 (LATE) 和AD有益。鉴于 NDD 通常具有多种蛋白病,预计 TDP-43 的鉴定将会与突触核蛋白一起受到重视。
(2)AD相关的混合神经病理在睡眠和蛋白质平衡中的相互作用
TDP-43 和 tau 是 FTD 和 ALS 中的主要蛋白病变,这两种疾病都存在睡眠障碍。在 ALS 患者的下丘脑中已经报道了兴奋神经元的丢失和 TDP-43包涵体产生。在松果体细胞和 SCN 血管活性肠多肽 (VIP) 神经元中观察到 C9orf72(ALS的常见遗传原因)扩增产生的二肽重复包涵体。这些调节昼夜节律的神经元不含磷酸化的 TDP-43 包涵体,表明 TDP-43 可能不是 ALS 相关睡眠缺陷的重要驱动因素,而 C9orf72,ALS 相关的运动和呼吸障碍可能是患者睡眠不足的更好指标。对于行为变异型 FTD,有证据表明食欲素失调与这些患者的睡眠障碍存在潜在关系。但类似对于 ALS,需要进一步的工作来描述 TDP-43、tau 和 FUS 病理学及其稳态对下丘脑功能和睡眠的作用。LATE和DLB是AD患者常见的混合病理,存在于 1/3至1/2的患者中。有趣的是,路易体病变(而不是TDP-43)与AD患者的睡眠障碍有关。鉴于RBD在PD和DLB等突触核蛋白病中的普遍存在,这些病理的存在可能会混淆将特定的Aβ 和/或 tau 驱动的病理分组为AD表型。但随着生物标志物的开发,这为阐明患者进展或恢复力的预测因素提供了令人兴奋的途径。
(3)睡眠生物检测:脑电图、多导睡眠图和可穿戴设备(活动记录仪和加速度计设备)
尽管早期干预和减少睡眠中断具有潜在益处,但睡眠中断的生物标志特征与 AD 和 PD 等 NDD 的风险仍未得到充分重视。睡眠障碍最可靠的生物标志特征是脑电图和睡眠阶段、休息和清醒周期的识别。进一步的日间睡眠,包括安静清醒的发作可能表明睡眠质量差。具有最高可溶性和沉积Aβ水平的大脑区域,如“默认模式网络”在安静清醒期间表现出高神经元活动。至关重要的是,在 NDD 和临床前研究中使用 EEG 是一种很有前途的方法,可以定义一系列 NDD(包括 AD、FTD 和 PD )的睡眠和认知功能障碍的预测生物学特征。新的可穿戴设备具有先进的睡眠检测功能,在监测 RBD方面很有用(包括PD或DLB前驱个体),可穿戴设备可以提供多种复杂的的测量指标,而脑电图识别微睡眠不稳定的效果更好。新的可穿戴设备也与 PSG/EEG 记录进行了比较,检测快速眼动睡眠的准确度为 0.51- 0.53,检测浅度睡眠的准确度为 0.52,检测深度睡眠的准确度为 0.79 - 0.83,表明这样的可穿戴设备仍然需要更高的准确性才能用作诊断工具。
(1)蓝斑-去甲肾上腺素能(清醒活跃,唤醒,抑制快速眼动)
过度磷酸化的tau和神经原纤维缠结在EC和海马积聚之前,更早影响的区域之一是LC(蓝斑)。LC 在 Braak 0 期和 I 期之间迅速积累 tau,到 Braak VI 期几乎所有剩余的神经元都含有 tau。Aβ 出现在 LC 的后期阶段,表明 tau 是 LC 损伤的选择性驱动因素。最近一份关于过度磷酸化的 tau (AT8 +) LC 神经元定位和形态报告表明 tau 从树突状扩散到 LC 相连区域的潜力(早在 Braak 0期),特别是从背侧 LC 到新皮质和海马。通过与丘脑、大脑皮层、基底前脑 (BF)、海马体和下丘脑的连接,包括抑制促进睡眠的 GABA 能神经元,LC 去甲肾上腺素能神经元促进唤醒和清醒并调节记忆,与AD 早期睡眠障碍相关。
去甲肾上腺素能信号发射在NREM 期间较低,在REM睡眠期间处于静止状态,这是由来自下丘脑外侧视前区 (VLPO) 和正中视前核以及下丘脑半胱氨酸(视前区 (POA))和黑色素浓缩激素 (MCH) 的 GABA 能抑制作用介导了神经元抑制。其他下丘脑输入包括食欲素、来自促进觉醒的组胺能神经元的间接激活,以及来自 SCN 通过下丘脑背内侧核的间接激活 。去甲肾上腺素能输出包括抑制 VLPO 神经元、促进食欲素介导的觉醒以及增加皮质锥体神经元兴奋性(图 2)。总而言之,LC 去甲肾上腺素能神经元赋予睡眠电路以神经调节作用,促进觉醒并将平衡转向清醒状态。去甲肾上腺素能对 tau 病理学和自噬失败的脆弱性,以及它在 AD 睡眠和昼夜节律障碍中的重要性,是 NDD 研究的一个新兴领域。
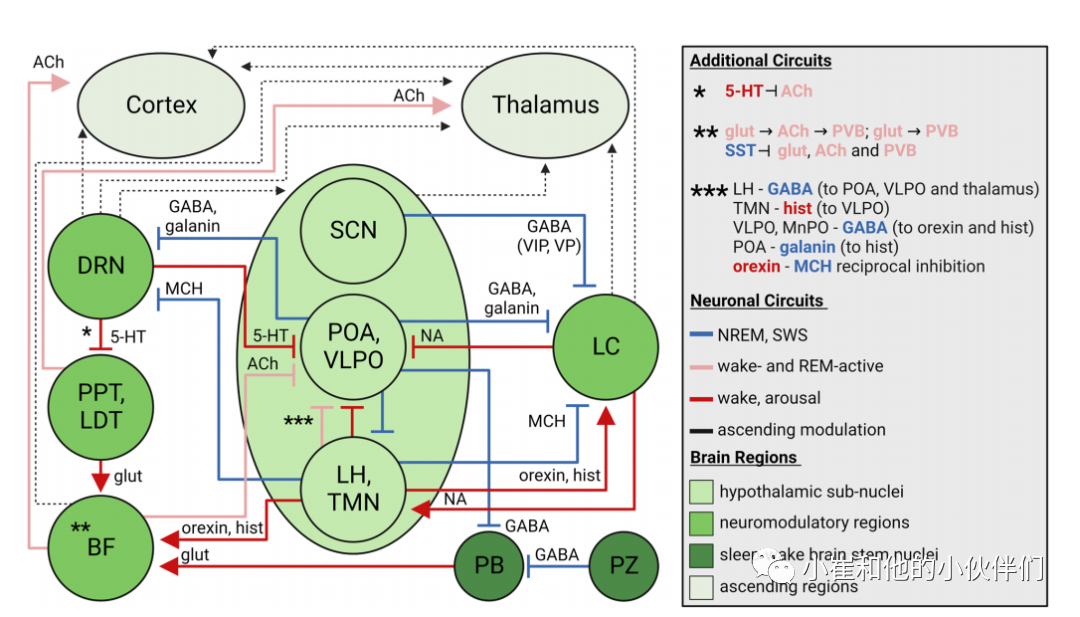
图2 睡眠-觉醒电路概述及其对非快速眼动、快速眼动和清醒状态的影响
(2)中缝背核 - 血清素能(通常促进唤醒和 REM 抑制,神经调节作用可以促进睡眠)
长期以来人们都知道血清素能信号可以控制睡眠-觉醒回路,在各种脑区和神经元群体中具有广泛的神经调节作用。血清素能神经元主要在背脊核(DRN)接收抑制性传入调节睡眠觉醒控制。血清素能神经元传出信号区域覆盖丘脑、下丘脑、大脑皮层、BF、其他脑干核团,还包括抑制REM的脑核和背侧被盖核团 (PPT/LDT) 的胆碱能神经元(图2)。这种活动将睡眠-觉醒平衡主要转向为清醒状态和 REM 抑制。重要的是,血清素活性在NREM 和REM 睡眠中降低,类似于观察到的去甲肾上腺素。总之,血清素是一种神经和睡眠调节剂,与下丘脑和胆碱能睡眠-觉醒回路密切相关。
(3)基底前脑,PPT/LDT-胆碱能(唤醒和 REM 活跃)和臂旁核-谷氨酸能(唤醒促进)
基底前脑(BF)是发挥广泛的神经调节作用的胆碱能神经元的主要中枢之一。这群神经元及其传出信号在 AD 中丢失是形成 AD 胆碱能假说和乙酰胆碱酯酶抑制剂治疗的核心原则。重要的是,BF 胆碱能神经元是睡眠-觉醒的调节器(以及调节其他行为,包括记忆和注意力),其较高的活动与清醒和 REM 睡眠相关,而 NREM 期间的活动较低 。皮质的广泛胆碱能神经间接激发锥体神经元,特别是与清醒和快速眼动睡眠密切相关的皮质-BF 振荡活动。这促进了高频皮层神经元活动,并抑制低频、慢 delta 波。胆碱能 BF 神经元的明显输入包括食欲素,其去极化或超极化取决于 5-HT 受体亚型的 5-羟色胺能活性,以及臂旁核谷氨酸能神经元的神经支配(图2)。臂旁谷氨酸能神经元促进觉醒,是脑干觉醒的主要来源 ,并且可以介导内感受相关的觉醒。其他谷氨酸能群也与睡眠-觉醒周期有关。臂旁核-BF-皮层回路对于促进觉醒状态至关重要(图 2)。
在BF 内,小清蛋白 GABA 能和谷氨酸能神经元也具有清醒和 REM 活性,并与局部胆碱能神经元相互连接。谷氨酸能神经元与胆碱能神经元和小白蛋白神经元形成突触,而胆碱能神经元直接与小白蛋白相连。这三个神经元群也分别被相邻的促睡眠生长抑素 (SST) GABA能神经元抑制,这体现了BF 电路作为睡眠调节器的复杂性(图 2)。
PPT/LDT胆碱能神经元的功能与BF中胆碱能神经元类似,对丘脑神经元有兴奋性传出。PPT谷氨酸神经元也支配 BF,并被证明在小鼠的化学基因激活后会引起广泛的觉醒,而在受到抑制时会引起更多的 NREM 睡眠(图2)。此外,PPT 胆碱能神经元的激活导致 NREM 中的 EEG 慢波减少,同时光/深 NREM 比率增加和 激活GABA 能神经元减少REM。
(4)下丘脑 – 食欲素能、组胺能(唤醒、唤醒活跃)和 MCH(促进睡眠、REM 活跃)
在下丘脑中,组胺能(结节乳头核)、食欲素能和 MCH(外侧下丘脑)神经元在 AD 中受到影响并积累 tau 病理。这些神经元网络是睡眠-觉醒周期的平衡唤醒(组胺能)、觉醒(包括基于食欲素的减少的 REM 和 SWS)以及从 REM 活跃的 MCH 神经元诱导睡眠的关键调节器,并接收来自神经调节性脑干核团(去甲肾上腺素、5-羟色胺和乙酰胆碱)的输入。在 AD 患者中,组胺能和食欲素能等促进觉醒的神经元丢失,但 MCH 神经元似乎得以保留并抵抗 tau 积累,表明下丘脑睡眠-觉醒控制失衡(特别是考虑到食欲素-MCH 相互抑制)。在 AD 患者中,食欲素能神经元减少,而 CSF 食欲素通常被报道为增加,表明一种复杂的,也可能是补偿性的(夜间睡眠不足和白天小睡增加)睡眠-觉醒信号失调机制。值得注意的是,啮齿动物的食欲能神经元随着年龄的增长而减少,老年大鼠和猕猴的海马和LC投射的食欲能神经支配分别减少。
啮齿类动物的睡眠限制会导致严重的细胞损失:LC、下丘脑、内侧前额叶皮层、CA1 和海马齿状回表现出神经元数量约 1/4–1/2 的明显损失。有趣的是,LC 去甲肾上腺素能神经元和下丘脑食欲素能神经元会随着慢性间歇性睡眠丧失而减少,但相邻的 MCH 神经元不会减少,并且所有 3 个神经元群都表现出轴突投射密度降低。这些影响在 4 周的恢复期后持续存在,表明这是一种持久的慢性过程,可能影响睡眠-觉醒周期的平衡,并可能在出现AD症状的个体中进一步加剧。
(5)下丘脑视交叉上核(昼夜节律发生器)
SCN是下丘脑前部的一个结构,它通过其他下丘脑核(如室旁下核和背内侧下丘脑核)的传入信号产生行为节律,然后传递给LC以影响唤醒。大多数SCN神经元是GABA能神经元,同时表达包括VIP和抗利尿激素等激素。分子钟和光刺激调节SCN神经元的活动,这些神经元在白天更活跃,在夜间受到抑制。重要的是,在阿尔茨海默病患者中,已记录到SCN神经元丢失和斑块病理学突出的缠结形成。并且SCN与包括AD、PD和HD在内的NDD的昼夜节律中断有关。鉴于SCN VIP神经元对C9orf72扩增的内含物的易感性,ALS和FTD也可能被考虑在内。
(6)GABA能回路(一般促进睡眠,NREM活跃)
最近的研究表明,除了来自单胺能神经元、胆碱能神经元和食欲素能神经元的调节之外,睡眠-觉醒周期的维持关键来自谷氨酸和 GABA 等快速神经递质。GABA 能中间神经元通过抑制作用促进整个大脑的睡眠,但在 AD 进展过程中这一作用受损 。VLPO 和POA 的中位视前核含有抑制觉醒促进的 GABA 能神经元,包括下丘脑(食欲素能和组胺能神经元)、DRN、LC 和臂旁核。从 POA 释放的半胱氨酸也可以抑制组胺能和去甲肾上腺素能神经元。对 VLPO 的输入包括抑制来自胆碱能神经元、去甲肾上腺素能神经元和较少的血清素能神经元以及组胺能神经支配的睡眠促进神经元。
重要的是,POA 和 VLPO 能特别强烈地启动睡眠,啮齿动物中 VLPO 的损伤导致约 40% 的睡眠时间损失。下丘脑中间核可能是 VLPO 的人类同系物,其中 AD 患者表现出半胱氨酸能神经元的丢失,神经元的数量与睡眠障碍的程度显著相关。在小鼠中,延髓的面旁区也被证明通过GABA能介导臂旁谷氨酸能神经元的抑制,是SWS和delta波脑电图的关键促进因子。还有一些非睡眠促进的GABA神经元,包括前面提到的BF小白蛋白神经元,以及下丘脑外侧GABA神经元,它们可以唤醒和REM活动,抑制促进睡眠的丘脑和POA/VLPO神经元。
在 Aβ 驱动的小鼠模型中,皮质 GABA 能张力的丧失与睡眠主导的慢波振荡活动的损害有关(减少功率而不改变振荡频率),并且 Aβ 的急性给药也能引起相同的电生理损伤。光遗传激活皮层兴奋性神经元拯救慢波振荡,恢复 GABA 受体水平,并且可以防止 Aβ 斑块的持续积累。这一数据表明 AD 中神经元回路、睡眠和神经病理学之间的动态相互作用。在小鼠睡眠剥夺期间,海马 SST 抑制性中间神经元被胆碱能和潜在的食欲素输入激活,从而抑制局部兴奋活动并损害记忆巩固 。然而,抑制性中间神经元表现出更高的自噬激活 BAG3水平,这暗示其可以防止 tau 积累。尚待阐明的是,SST 自噬恢复力以及 AD 中对 Aβ 和 tau 的脆弱性与长期清醒发生的增强抑制门控有关。
总的来说,这些睡眠调节区域和神经元在睡眠-蛋白质稳态相互作用中非常重要,因为它们对蛋白质病的不同脆弱性或恢复力会影响睡眠-觉醒平衡的调节,因此可能会促进 AD 进展和蛋白质积累。
睡眠是一种广泛的多细胞现象,由具有细胞/分子水平调节的稳态过程调控,其中分子的积累与清醒的持续时间成正比。大量的此类分子和蛋白质的不良数量会随着清醒持续时间的延长和睡眠的中断而积累 ,从而导致 NDD 的病理扩散。
(1)一夜睡眠不足会增加健康成年人的阿尔茨海默病相关蛋白病
Aβ和tau在正常睡眠后的CSF中早晨较低。然而在没有认知障碍的中年人(年龄范围:40-60 岁)中,即使限制睡眠一晚后,其清除率也会受损(表现为 Aβ42 水平升高)。相反,清晨血浆中的总 tau 蛋白比晚上的水平增加了 1.8%,而且令人惊讶的是在年轻男性(约 22 岁)睡眠剥夺一夜后其增加了 17.8%,但血浆 Aβ 没有变化。此外,在健康成人(年龄范围:22-72 岁)失眠一夜后Aβ18F-florbetaben PET 信号增加(特别是海马体中)。Holth和 Lucey及其同事的研究表明,健康成人(30-60 岁)在一夜睡眠不足的情况下CSF Aβ 增加约 30%,CSF tau 增加 > 50%。
在健康成人(年龄范围:20-40 岁)连续五晚部分睡眠剥夺后,观察到 CSF 食欲素浓度增加了 27%,而淀粉样蛋白、星形胶质细胞或神经变性生物标志物没有变化。值得注意的是,除了 SWS外所有睡眠阶段的时间都减少了。这种睡眠仅被部分剥夺时残留的 SWS可能会促进蛋白质清除,防止淀粉样蛋白增加(在完全急性剥夺可以观察到)。
(2)淋巴清除
成人(平均年龄约 42 岁)一晚完全睡眠剥夺后,大脑对磁共振成像示踪剂的清除率立即降低,甚至在睡眠恢复后仍持续。这种剥夺可能减少了由神经元活动调节的淋巴系统清除率,并在 SWS 期间增强 。淋巴系统清除是大脑中间质 (ISF) 清除的两种机制之一,也是睡眠-蛋白质稳态轴和 Aβ 和 tau 清除中需要考虑的关键机制。
淋巴系统清除是大量液体交换的途径,其中 CSF 从蛛网膜下腔泵入大脑,首先进入皮质软脑膜动脉。动脉血管动力将 CSF 移动到动脉周围 Virchow-Robin 空间的更深脑区,其穿过血脑屏障 (BBB) 的运输由包裹脑血管系统的星形胶质细胞末端上的 AQP4 通道介导。CSF-ISF 交换后,液体沿静脉周围空间流出至硬脑膜淋巴系统,促进脑细胞外代谢物和溶质的清除。在 SWS 期间,间质空间增加 60%,支持更高的淋巴系统介导的 Aβ 清除率;但在清醒或睡眠剥夺期间清除率降低,小鼠的 ISF tau 增加 90-100%(图 3a)

图3 .睡眠不足破坏了Aβ和tau蛋白的平衡,导致蛋白质病变、神经网络功能障碍和认知障碍
这些结果表明淋巴系统介导的 Aβ 和 tau 清除与睡眠的密切关系,以及在 AD 患者睡眠丧失时可能发生的正反馈回路。AD 中会出现大量的血管损伤,包括脑淀粉样血管病、血管迂曲和僵硬、脑血流减少以及动脉周围和静脉周围间隙扩大。所有这些都会损害 淋巴系统清除,并且由于大量 Aβ 和 tau 导致淋巴系统清除失败而进一步受损。有趣的是,大鼠大脑中淋巴系统清除率的昼夜波动显示出区域差异,在涉及昼夜节律调节的大脑区域(包括 SCN 和外侧下丘脑)与睡眠相关的清除大量增加。关键的是,急性睡眠剥夺会显著增强 Aβ 和 tau,这会进一步损害睡眠并导致蛋白质清除率持续受损 。
蛋白质降解的机制,特别是自噬-溶酶体途径 (ALP)、泛素蛋白酶系统 (UPS) 和未折叠蛋白反应 (UPR) 在功能上位于细胞内。然而,这并不排除这些过程对睡眠的影响,因为神经元群体或大脑区域的蛋白质稳态受损会产生网络效应 。例如,有学者证明了人脑组织 EC 兴奋性神经元对自噬缺陷的细胞和区域特异性,促进了它们对 tau的脆弱性。最近,神经元自噬溶酶体酸化失败被确定为AD 患者和小鼠模型中Aβ斑块形成的先兆。这些神经元的自噬受损影响可能会导致病理蛋白在网络层面扩散(包括在睡眠中断期间)。Aβ 和 tau 对神经元网络功能障碍的协同作用以及蛋白病变在细胞间的传播进一步加剧了整个大脑中蛋白质稳态失衡的影响。因此,睡眠和昼夜节律与自噬、UPS 和 UPR 的相互作用可能是驱动早期 AD 阶段Aβ 和 tau 蛋白病的关键机制。图 3 显示了AD中睡眠与细胞蛋白质稳态机制相互作用的相关性(图 3b),以及睡眠病理学如何影响神经元回路(图3c)和认知能力下降(图 3d)。
(1)自噬和睡眠障碍
AD 中自噬通量紊乱的最早特征之一是大量自噬体,它们积聚在神经元细胞质、轴突中,最明显的是在营养不良的神经元中。自噬体无法清除会阻碍Aβ、tau 和其他蛋白质的降解。随着自噬缺陷的增加,神经元损伤也会增加,从而导致 AD 进展 。此外,未清除的自噬体和自噬溶酶体通过外泌体释放加速 Aβ 和 tau 的细胞间传播 。有趣的是,扰乱睡眠会重现 AD 相关的自噬功能障碍,而敲除自噬功能足以重现神经退行性表型 。
果蝇中自噬通量的遗传操作证明睡眠和自噬之间的双向联系。在睡眠期间,自噬体比清醒期下降。阻断自噬体形成可增加睡眠,而阻断自噬溶酶体降解可减少睡眠 。在小鼠中,急性睡眠剥夺导致自噬体在海马神经元中积聚,LC3B、Beclin-1 和 p62 的表达增加,表明自噬过程的募集 。慢性睡眠片段化小鼠的自噬通量被破坏,自噬体、内体以及细胞内溶酶体的数量和大小增加。这伴随着急性和慢性睡眠剥夺后的空间学习和记忆障碍。因此,没有溶酶体融合和蛋白质降解的自噬募集会随着睡眠障碍的增加而发生,并进一步促进蛋白质病变和疾病进展(图 3b,ALP)。
有趣的是,有研究报告了慢性睡眠片段化野生型小鼠的皮质和海马 Aβ 细胞内积累,类似于 AD 模型中的观察的结果。作者将此归因于正常淀粉样前体蛋白 (APP)通过内体-自噬体-溶酶体 (EAL) 途径的加工失败,其中通过 EAL 途径的流量减少会损害APP 运输和清除,从而促进淀粉样蛋白形成和 Aβ 积累。
(2)自噬的昼夜节律
昼夜节律和睡眠-觉醒周期影响基因表达并与 AD 病理学相关。AD 患者表现出昼夜节律失常,在临床上通过活动/休息周期测量,表现出周期阶段延迟和较低的振幅峰值。在敲除生物钟基因 Per1 的老年小鼠中,自噬受损,Aβ42 和早老素水平较高 。Per 基因(包括 Per1 和 Per2)编码 PER1 和 PER2 蛋白(通常统称为 PERIOD 蛋白),是昼夜节律振荡时间的重要调节器。更重要的是,生物钟调节自噬基因的表达(包括促进自噬激活节律表达的转录因子 Nr1d1 和 C/EBPβ以及 Beclin-1 和 atg4a )并触发自噬体形成。在小鼠海马体中,与自噬相关的 LC3-II (而不是细胞质 LC3-I) 表现出昼夜节律性,表明与睡眠相关的光照阶段自噬体形成和自噬通量比黑暗阶段更高。这与 AD 中自噬通量和蛋白质清除失败造成的自噬体过度积累不同。值得注意的是,ALP 是按昼夜节律周期调节的,AD 中的心律失常可以进一步加剧已经存在的自噬缺陷,类似于睡眠障碍发生的情况(图 3b,ALP)。
在小鼠慢性睡眠打断后,海马体自噬通量的整体光相峰值和暗相低点仍然存在,但节律性受损,每个阶段内都有异常变化。急性恢复睡眠不足以逆转这些变化,并仍会在睡眠相关的光照阶段触发 Beclin-1 表达增加。从这项工作中可以得出两个结论来构建对 AD 中睡眠-蛋白质稳态相互作用的理解。首先,睡眠不足会扰乱自噬通量的昼夜节律调节,导致 Aβ 和 tau 聚集增加。其次,这些缺陷在恢复后仍然存在,包括 Beclin-1 对 AD 中已经不堪重负的自噬溶酶体系统的持续、异常激活,表明 AD 进展的慢性并发症。
(3)未折叠蛋白反应 (UPR) 会随着年龄、睡眠不足和阿尔茨海默病发生而受损
UPR通常负责清除错误折叠的蛋白质, 但在 NDD 中不堪重负,并且在睡眠中断时进一步恶化。蛋白激酶 RNA 样 ER 激酶 (PERK)、肌醇获取酶 1α (IRE1) 和激活转录因子 6(ATF6α 和 β)是帮助启动 UPR 的三种关键蛋白,在应对错误折叠的蛋白质时结合免疫球蛋白 (BiP) 并从中解除结合。UPR 的慢性激活,特别是在 PERK 分支上,发生在 AD 患者和其他 tau 蛋白病的大脑中。AD大脑具有更高的 UPR 激活标记物表达,包括磷酸化 PERK、真核起始因子 2α (eIF2α) 和 IRE1以及 BiP。Tau 病变的 UPR 增加,与这些疾病中的早期海马 tau 病理学相关。此外,显示异常 tau 磷酸化水平的神经元和神经胶质细胞也展示出相应的 UPR 激活标志物增加,证实了它们之间的联系。
UPR 伴侣在清醒期间或睡眠不足时上调,并通过清除错误折叠的蛋白质和减少蛋白质翻译来帮助缓解内质网 (ER) 压力。ER 伴侣 BiP 与错误折叠的蛋白质结合以防止聚集并促进重新折叠。在小鼠皮层中,BiP 水平随着睡眠被剥夺而逐渐增加,并作为对未清除蛋白质积累的补偿。这种现象在果蝇中也被观察到,其中 BiP 水平在睡眠不足时升高,并在恢复睡眠期间下降至基线水平。增加正常的 BiP 表达,或增加降低 BiP 功能的阴性形式,分别延长或减少睡眠剥夺后的睡眠恢复。BiP 过表达会减慢 UPR 功能,这表明 AD 患者的慢性睡眠中断导致 UPR 被大量错误折叠的蛋白质淹没的状态(图 3b,UPR)。并且这些遗传学操作对果蝇的基线睡眠没有影响。
有趣的是,年轻小鼠的急性睡眠剥夺促进了蛋白质清除并通过 UPR 减少了翻译,但在老年小鼠中却导致了促凋亡信号。睡眠和蛋白质质量控制都会随着衰老而受损,导致 UPR 中再折叠方面的效率降低 。从年龄相关疾病和老年野生型啮齿动物中伴侣蛋白供应不足可以看出这一点,证实了睡眠质量和蛋白质稳态相互影响。UPR 在食欲素能和去甲肾上腺素能唤醒活性神经元中被激活,磷酸化 PERK 增加。这在老年小鼠中发生的程度更大,与食欲素能和去甲肾上腺素能神经元活动的下降以及 CCAAT/增强子结合蛋白同源蛋白 (CHOP) 的核转位/激活相关。已知 CHOP 会发出细胞凋亡信号以响应 ER 应激并介导睡眠呼吸暂停/缺氧相关的细胞应激和损伤。
蛋白质稳态系统的应激反应和质量控制在老年候选人的几乎所有组织中都变得功能失调,这表明AD 和其他神经退行性疾病的复杂相互作用,其中年龄、蛋白质稳态受损和睡眠对疾病进展产生单独和协同影响。重要的是,UPR 伴侣介导的治疗可恢复小鼠衰老相关的睡眠和认知障碍。NDD 的另一个 UPR 治疗靶点是 eIF2α,当它被磷酸化的 PERK 磷酸化时,它会减弱对记忆和神经元功能至关重要的全局蛋白质合成率。磷酸化 eIF2α 水平在阿尔茨海默病和其他 NDD 中升高。临床适用疗法的体外筛选试验发现,抗抑郁药盐酸曲唑酮和另一种化合物(二苯甲酰甲烷)逆转了磷酸化-eIF2α 对蛋白质合成的影响。这两种化合物的体内治疗使翻译正常化,并对朊蛋白和 tau 病模型具有神经保护作用。重要的是,曲唑酮可用于治疗失眠和改善睡眠维持,进一步表明睡眠-蛋白质稳态相互作用的治疗潜力。
UPR 的持续上调在广泛的蛋白质病和睡眠丧失后是有害的,它中断蛋白质合成,促进神经变性并加剧有缺陷的睡眠蛋白质稳态正反馈回路。如果蛋白质不能在 ER 腔(蛋白质合成和包装位点)中适当折叠,蛋白质将被引导到蛋白酶体以避免聚集。最后,温和 ER 应激可以通过募集自噬过程为 UPR 的神经保护机制提供前提条件。然而,随着慢性睡眠剥夺、UPS 和 ALP 募集等长期压力以及补偿 UPR 失败,可能会进一步压倒细胞蛋白稳态。
(4)泛素蛋白酶体系统 (UPS) 和睡眠障碍
UPS 和睡眠中断之间的相互作用不像自噬和 UPR 那样明确。有证据表明,OSA 通过间歇性缺氧降低蛋白酶体活性,这会导致神经变性后蛋白酶体功能障碍。随着 UPS 缺陷的增加,PER1 和 PER2 蛋白、生物钟蛋白和昼夜节律调节器的泛素化和降解变得不平衡,延长了昼夜节律。Tau 积聚与蛋白酶体功能障碍密切相关。尽管需要研究来阐明是否存在确定的 tau-UPS-睡眠成分导致 AD 中的蛋白质病和睡眠丧失。重要的是,UPS 的功能障碍导致自噬过程的补偿性激活,而自噬在 AD 中已经异常,这将导致睡眠-蛋白稳态轴的慢性缺陷。
(1)疾病模型:阿尔茨海默病蛋白病通过神经元活动传播
近年来, AD蛋白病传播是神经元活动的反作用的假设越来越受到关注。几个小组使用不同的实验模式得出了相同的结论。从机制上讲,神经元活动促使tau 释放到细胞外空间,在那里它可以通过细胞间传播而扩散和播种病理(对于 Aβ 也观察到了这一点)。在人类中,较高的海马激活与 Aβ PET 水平呈正相关,并与记忆力下降纵向联系 。
在 SWS 期间神经元活动最受抑制。因此,AD 患者的睡眠不足、觉醒增加和清醒时间更长导致神经元高兴奋性持续时间更长,以及增加 Aβ 和 tau 扩散。NREM 慢波活动的中断与认知健康老年人的内侧前额叶皮质中 Aβ 水平的增加成正比。同样,tau 的增加与 delta 功率的降低 (1–4 Hz)相关。这些研究证实了睡眠、神经元回路中断以及 Aβ 和 tau 之间存在联系(图 3c)。在健康人的 SWS 期间特异性诱导觉醒时,早晨 CSF Aβ 水平升高可能是由较高神经元活动期间 ISF Aβ 增加(以及细胞外 tau 释放)驱动的。ISF Aβ 浓度在清醒期间较高,在睡眠期间较低。年轻 Tg2576 小鼠海马ISF Aβ 和乳酸水平在黑暗期间比光照期增加 > 20%,这是神经元活动的标志。
抑制性神经元在减弱的深度睡眠状态下占主导地位。脑干神经元平衡 NREM 和 REM 之间的相互作用(主要是 GABA,有一些证据表明是谷氨酸能调节器),表明 SWS 和 NREM 睡眠对于切换到 REM 状态的重要性。血清素能和去甲肾上腺素能的张力在NREM 中减弱,并在 REM 睡眠中静止。血清素和去甲肾上腺素的抑制,包括来自下丘脑 MCH 神经元和 POA GABA能和半胱氨酸神经元的抑制,也与 REM 睡眠的出现和维持有关。较高的神经活动率与APP处理增加有关,而神经退行性环境中的慢性应激源(即氧化应激)可以进一步将平衡转向淀粉样蛋白生成与非淀粉样蛋白生成处理。有证据表明睡眠障碍期间 Aβ 产生增加,会导致健康成人(年龄范围:30-60)的 CSF Aβ 水平升高。因此,随着深度睡眠和快速眼动睡眠的丧失,Aβ 生成率增加可能是将蛋白质病与睡眠和神经元活动联系起来的额外机制,尤其是REM在睡眠期间通常处于低活动状态的去甲肾上腺素能和血清素能神经元中。
在 EC 中过度表达人类 APP 和 tau 的AD 啮齿动物模型中,Aβ 的存在加速了 tau 的积累并扩散到海马体,并导致 EC 兴奋性神经元过度活跃,放电率更高。阻断这些高放电率的神经元活动随后会抑制 Aβ 和 tau 的积累和扩散。这项工作提出了一种疾病模型,其中 Aβ 诱导的过度兴奋性增强了 tau 错误折叠和聚集,导致 tau 诱导的神经变性和神经元沉默。高频率的神经元活动(如AD中睡眠障碍)会降低信号、噪声,从而导致非特异性和潜在异常的突触连接和可塑性。这构成了睡眠-突触稳态假说的一部分,该假说指出,睡眠休息质量来自于维持突触能量使用、压力、代谢需求、可塑性和活动之间的平衡,从而促进健康的神经元和认知功能。
最近研究证明,神经元活动与BBB 流出受体和内皮细胞的核心生物钟基因表达呈负相关,包括 PAR bZip 和 Bmal1 依赖信号。作者提出清醒状态下神经元活动依赖性抑制BBB 流出,而在睡眠状态下则增强。当睡眠受损和过度兴奋发生时,这可能导致 AD中 通过这些机制清除 Aβ 失败。鉴于神经元活动在影响蛋白病方面的重要性,最近的一项研究表明,与非转基因同窝小鼠相比,APP 转基因小鼠在丘脑网状核 (TRN) 的神经元活动减少,导致睡眠碎片化增加和 SWS 减少。使用兴奋性 DREADD 选择性激活 TRN 可以挽救上述缺陷,从而挽救海马体和皮质中的淀粉样斑块负荷。
总的来说,这些报告表明神经元活性可以调节Aβ 和 tau产生、扩散、清除和相互作用,进而促进Aβ 和 tau积聚并引发AD的神经网络功能紊乱和认知能力下降。睡眠表现出明显的神经元电生理特征,包括NREM 睡眠期间 UP 状态丘脑纺锤波 ( 10–15 Hz) 和海马锐波波纹 (100–250 Hz) 和 DOWN 状态皮质三角波 (1–4 Hz)和 K 复合波(低频、高振幅)以及 REM 睡眠期间的海马 theta 振荡(4-10 Hz)之间的适时切换。睡眠相关神经元活动的协调和时间增加了信噪比,加强了与记忆相关的突触连接(通常是新形成的),并有助于记忆巩固(图 3d)。总之,这些结果表明调节神经元活动,特别是与睡眠相关的电生理学,是改善认知和促进蛋白质稳态的有效AD治疗策略。
(2)曲唑酮
盐酸曲唑酮是一种独特疗法,因为它在睡眠-蛋白质稳态轴上靶向多个目标。曲唑酮是一种被批准用于治疗重度抑郁症的抗抑郁药,通常用于睡眠障碍患者和 AD 患者的超适应症治疗。多种神经递质系统受到曲唑酮治疗的影响,包括 5-羟色胺受体的选择性激动和拮抗作用、5-羟色胺转运体抑制和再摄取抑制、组胺能受体和 α1 和 α2 肾上腺素能受体的拮抗作用,以及在较小程度上表现出抗胆碱能活性。总之,曲唑酮的神经调节作用可以使神经元睡眠-觉醒平衡并朝向睡眠(图2和4)。
在一项3期临床试验中,患有睡眠障碍的AD患者在2周的时间内每天睡前服用50mg曲唑酮。治疗显著增加了睡眠时间,并倾向于减少整个晚上的觉醒和觉醒时间,而不会加剧白天小睡或EDS。通过这种短期治疗,认知能力不受影响。有证据表明在4年的随访中,伴有睡眠障碍的AD患者长期使用曲唑酮(中位剂量:50 mg/天)可减缓最小状态检查的认知能力下降。在200mg的高剂量下,曲唑酮在AD患者中是安全可行的,并且在调节UPR的预测范围内。虽然曲唑酮-UPR相互作用机制是通过使蛋白质合成正常化,但Aβ 和 tau降解和清除可能通过减轻神经退行性负担间接改善,并减少UPR过度激活改善细胞和蛋白平衡。
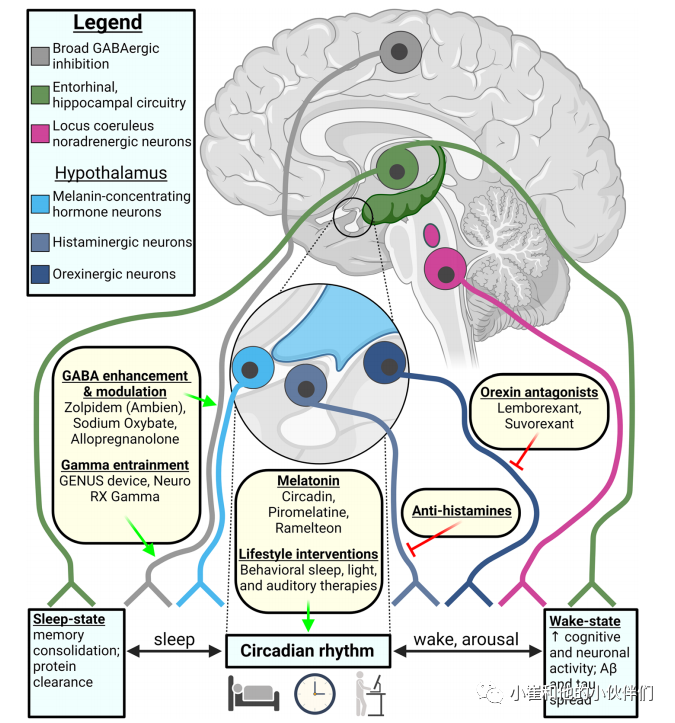
图4阿尔茨海默病睡眠-觉醒周期的神经元控制和睡眠恢复的治疗靶点
(3)肾上腺素能α受体激动作用
肾上腺素α受体激动剂通过降低蓝斑去甲肾上腺素能活性、降低觉醒和促进睡眠来影响睡眠-觉醒周期。右美托咪定是一种α2肾上腺素能受体激动剂,在体外和体内临床前模型中显示出治疗效果(包括抗炎、促进认知和促进神经营养作用),但在治疗后细胞和啮齿动物中tau磷酸化增加(即pS202、pT205、pT231、pS396)持续长达6小时。由于右美托咪定可诱导NREM 2- 3期睡眠(包括增加脑电图慢波δ振荡和纺锤波),因此其在临床上被用作镇痛和镇静剂。右美托咪定在睡眠-蛋白平衡相互作用中的一个潜在好处是它可以急性增强淋巴细胞,但作为AD主要治疗药物因其加剧tau病理使用仍然受限。
(4)食欲素拮抗剂——Suvorexant
目前,AD 中最有希望的睡眠调节疗法是食欲素拮抗剂 suvorexant,它被用于治疗失眠,并于 2020 年 2 月获批用于治疗 AD 患者的睡眠障碍。据报道,食欲素信号也会对认知产生影响,在 AD 中存在食欲素紊乱和CSF 食欲素升高。Suvorexant 与食欲素竞争结合受体 OXR1 和 OXR2从而促进睡眠。在治疗轻度至中度 AD 失眠的 3 期试验中,每天口服 suvorexant 治疗 4 周后总睡眠时间比基线显著延长 73 分钟(安慰剂组为 45 分钟),并且治疗不会恶化认知并且耐受性良好。最近Lucey 及其同事证明,对健康的中年成人进行急性 suvorexant 治疗可使 CSF 的Aβ 水平降低 10-20%,p-tau (T181) 水平降低 10-15%。Suvorexant 可能在联合疗法中也是有益的,可能与最近批准的 aducanumumab 或 lecanemab 一起使用,并探测睡眠-蛋白质稳态轴。在临床前 AD 模型中使用 suvorexant 促进睡眠可用于评估特定神经元对 tau 和 Aβ 易感性的改变,以及 淋巴清除率和自噬通量的潜在改善。食欲素功能的调节虽然仅限于小细胞群,但可以通过促进睡眠帮助平衡昼夜节律失常从而减轻蛋白质稳态的有害变化,进而在整个大脑中发挥有益的网络效应。重新平衡特定的神经元群体功能可能会纠正 AD 中出现的全网络功能障碍。例如,suvorexant 治疗可增强 AD 模型小鼠的海马长时程增强作用。
在睡眠和 AD 相关神经元和区域、下丘脑食欲素神经元以及 EC-海马认知回路(图 4)中表征 suvorexant 治疗对蛋白质稳态的改善,将有助于确定睡眠和蛋白质稳态在疾病进展中的相互作用,并根据疾病分期提供治疗效果。
本文作者探讨了睡眠-蛋白质稳态轴的实验对于理解 AD 进展和年龄的关联的重要意义。未来的工作需要阐明前驱 AD 的新标志物,包括 PET 和血浆生物标志物以及指示性EEG,以显示神经元对蛋白质稳态改变的脆弱性和睡眠中断。此外,可以针对睡眠和蛋白质稳态测试单一疗法和组合疗法,包括其疗效在 AD 进展中的变化。值得注意的是,睡眠障碍通常与下丘脑-垂体-肾上腺轴活动和行为压力同时发生,这也会对细胞蛋白质稳态产生影响。在临床前睡眠剥夺范式的设计和解释中应考虑减轻压力,以减轻蛋白质稳态中与压力相关的额外改变。探测神经元电生理和睡眠模式将允许识别和重新利用针对这些机制及其与蛋白酶平衡的相互作用的治疗方法,从而促进新药的发现。睡眠障碍、蛋白质病变和蛋白质平衡功能衰竭是衰老和NDD中常见的现象,因此本文提出的结论适用于广泛的脑部疾病,包括混合病理的情况。

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言