最新Circulation:消除衰老细胞可促进肺动脉高压的发展和恶化
2022-12-18 肺动脉高压研究进展 肺动脉高压研究进展 发表于安徽省

解除衰老的内皮细胞的影响可能会破坏对肺血管细胞生长的控制,从而促进或加剧肺动脉高压的发展或进展。衰老干预在许多情况下被证明是有益的,但在使用时应注意其对内皮细胞和肺血管的潜在影响。
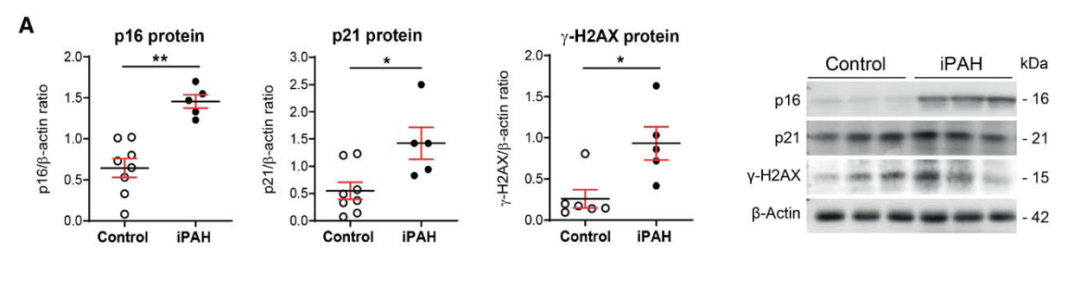
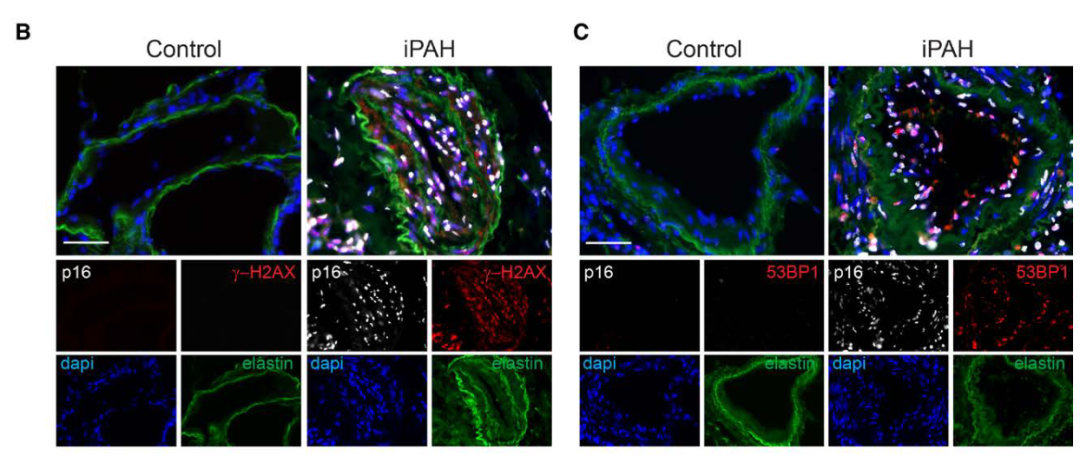
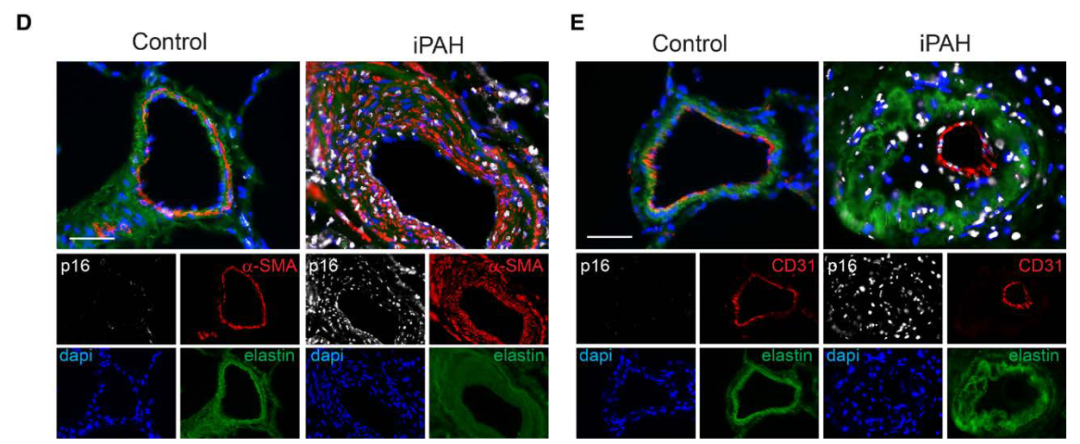
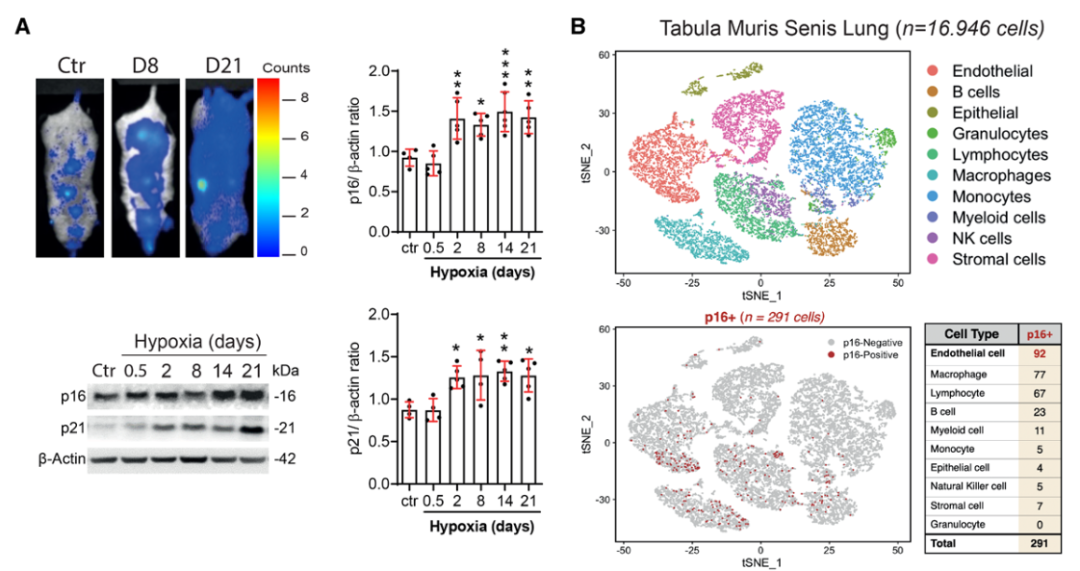
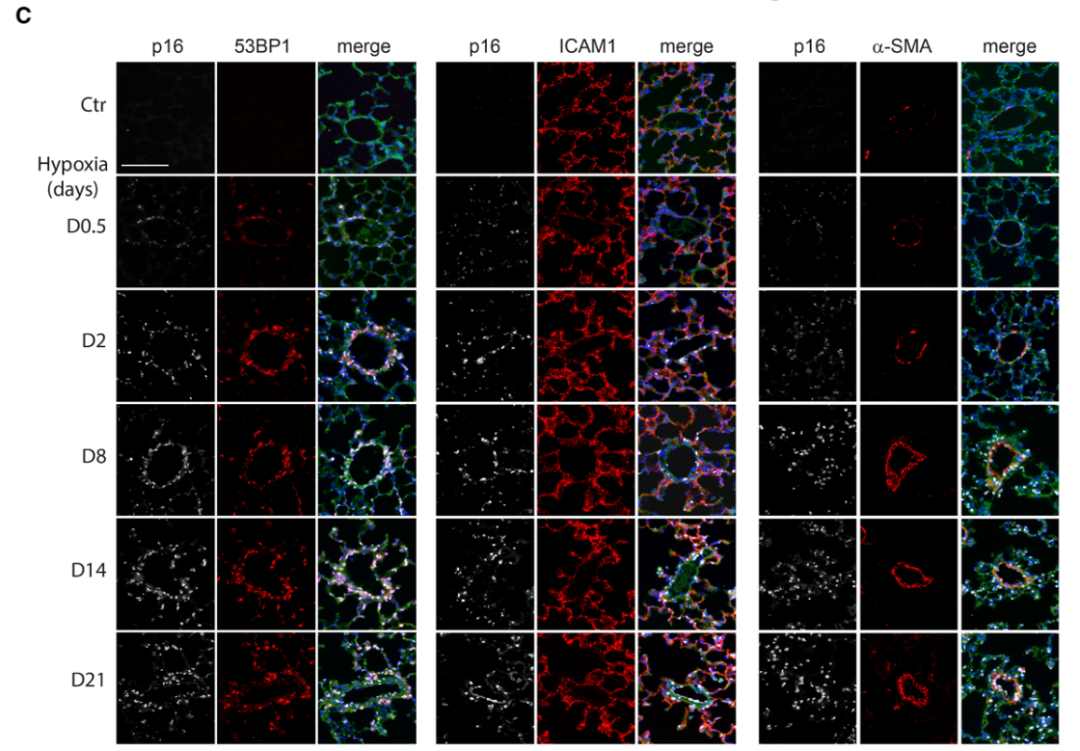
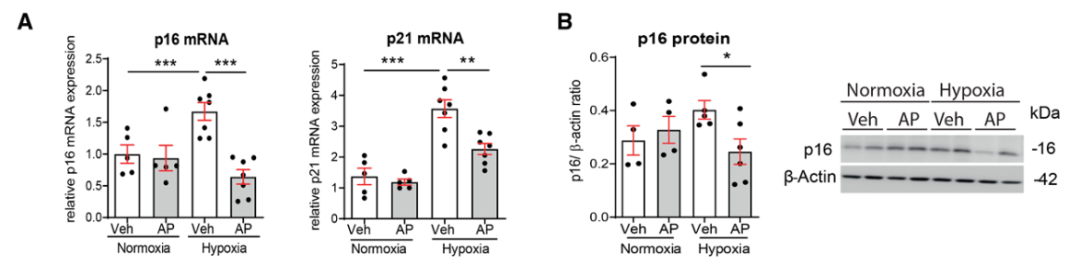
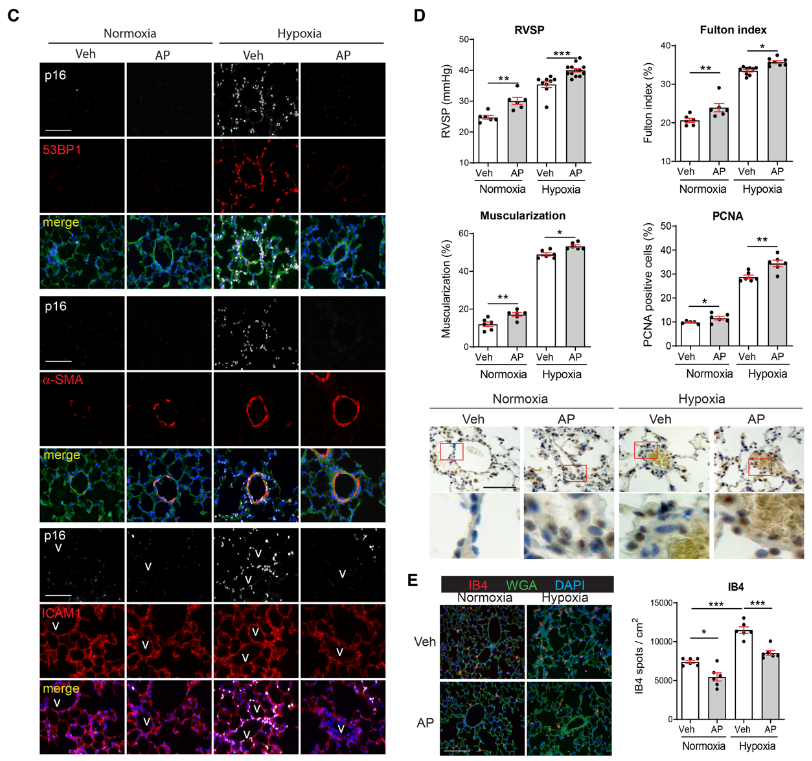
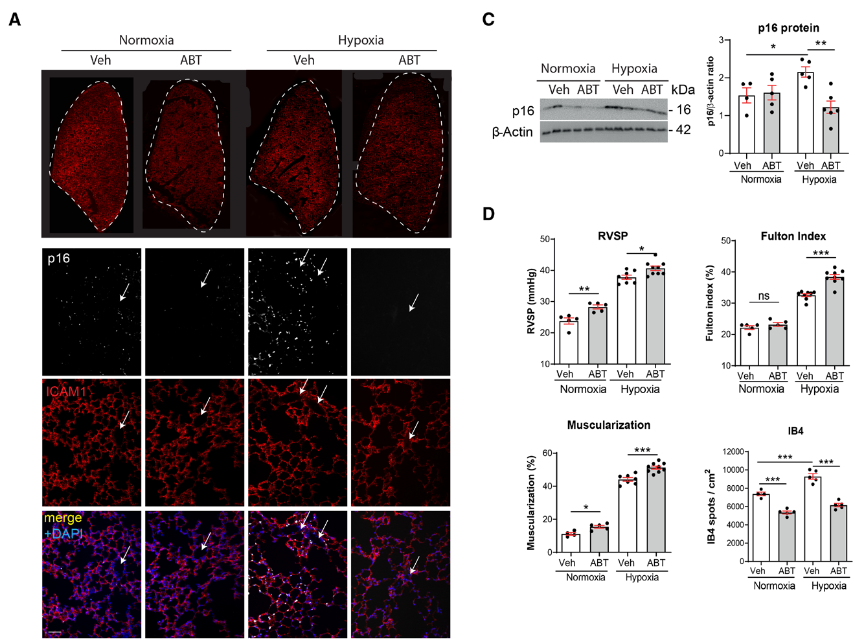
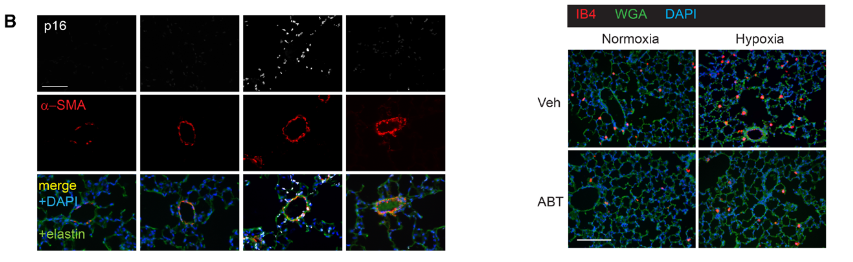
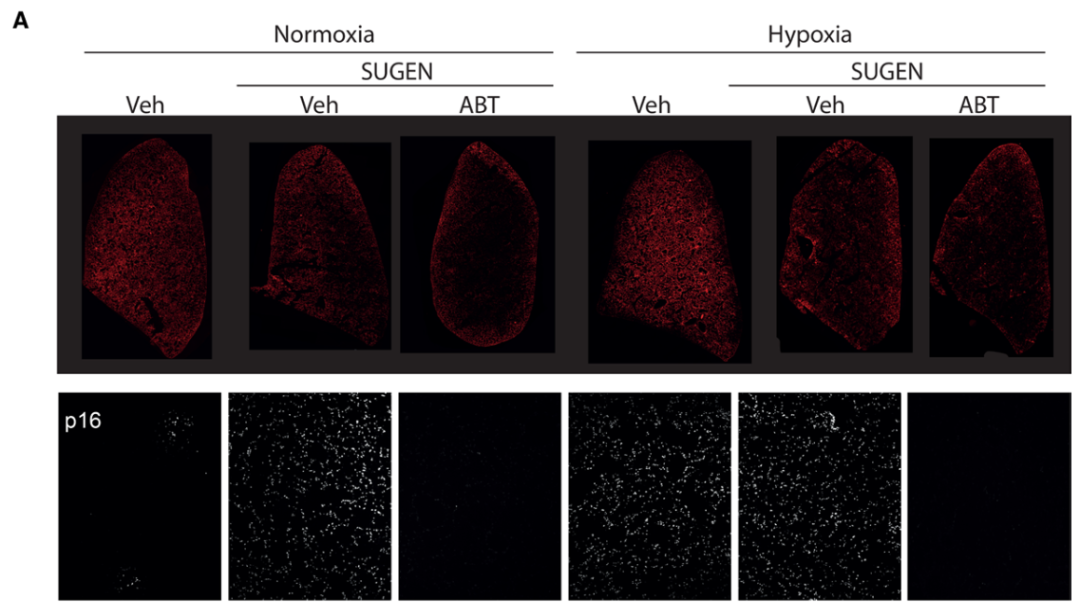
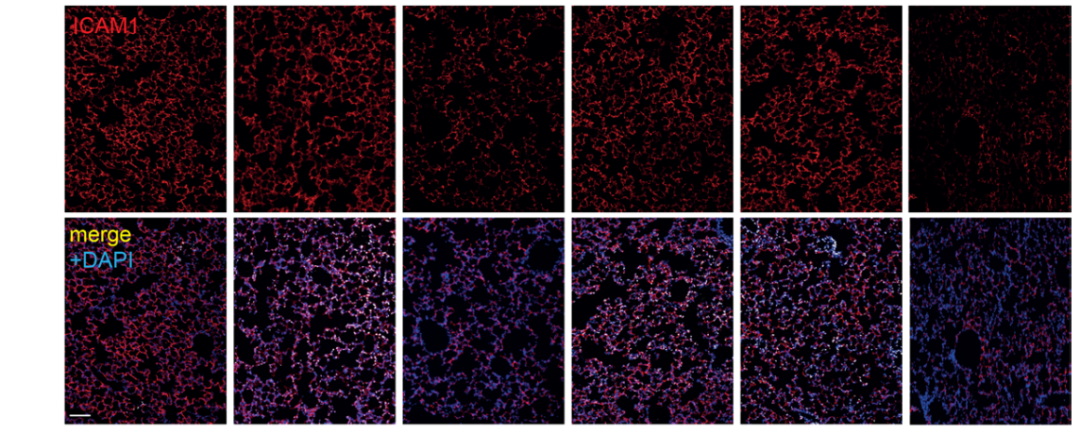
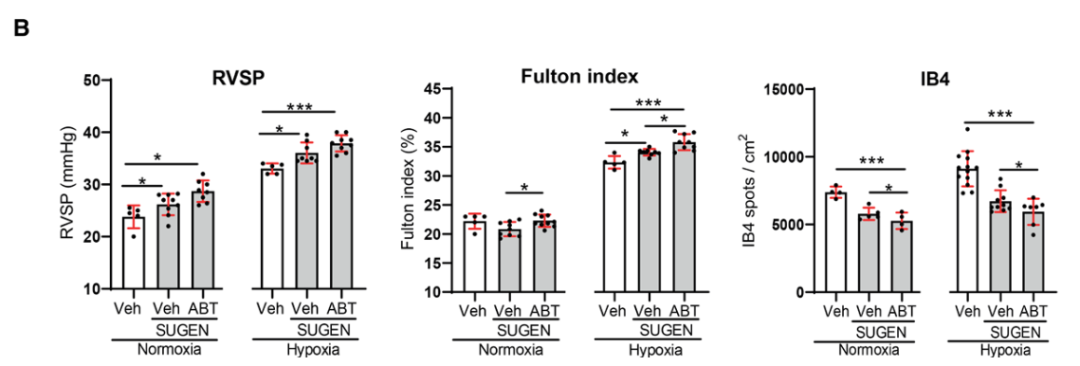
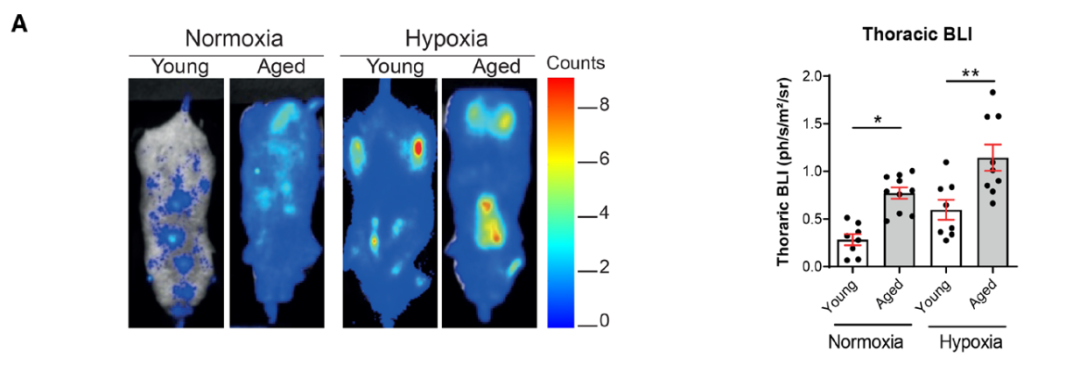
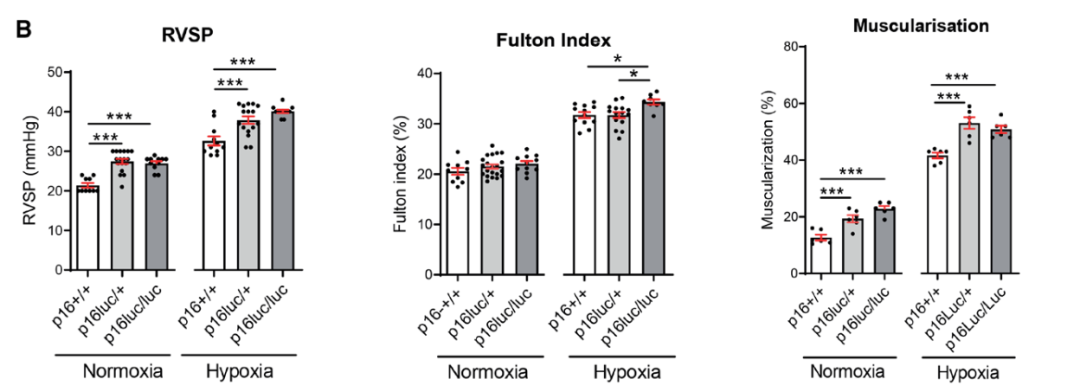
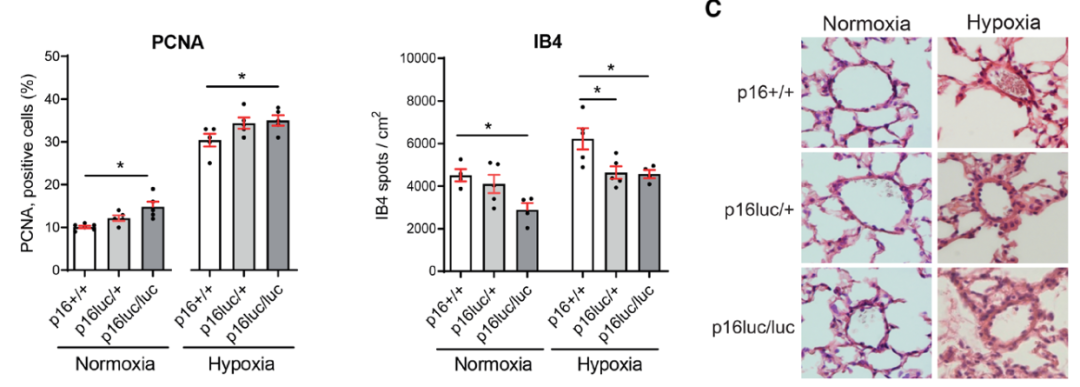
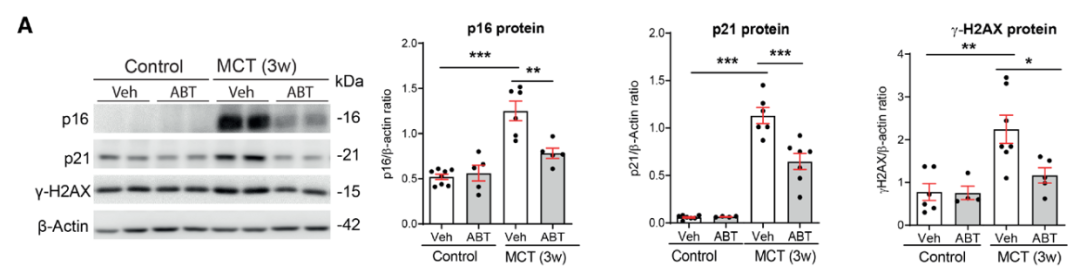
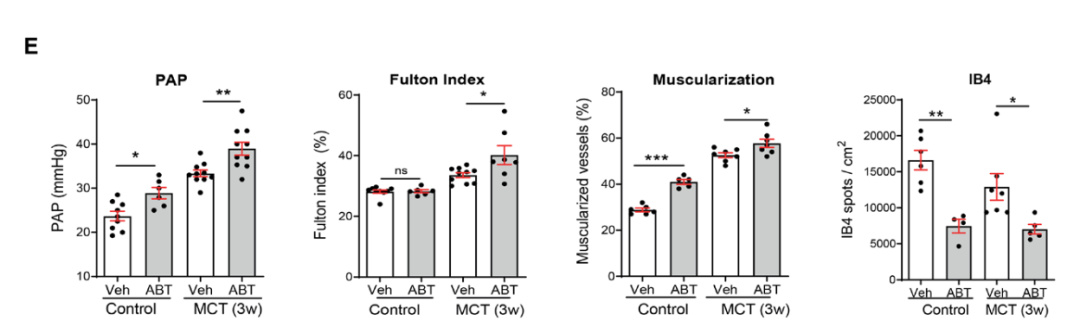
研究背景 衰老细胞(SCs)参与增殖性疾病,但它们在肺动脉高压中的作用仍未确定。该研究分析了肺动脉高压患者的SCs以及SCs在动物肺动脉高压模型中的作用。 研究方法 该研究检测了肺动脉高压患者和小鼠模型中的衰老(p16, p21)和DNA损伤(γ-H2AX, 53BP1)标志物。在p16-荧光素酶(p16LUC/+)敲入的小鼠中通过发光成像监测p16的激活。通过自杀基因(p16-ATTAC小鼠中p16启动子驱动的杀手基因构建体)、衰老药物(ABT263和细胞渗透性FOXO4-p53干扰肽[FOXO4-DRI])以及p16LUC/LUC小鼠中的p16失活获得SC清除。暴露于常氧、慢性缺氧或缺氧+Sugen的小鼠、过量表达5-羟色胺转运体(SM22-5-HTT+)的小鼠以及给予野百合碱的大鼠的肺动脉高压。 研究结果 第一,细胞衰老与特发性PAH患者的肺部血管重塑有关。与对照组相比,肺动脉高压患者的肺部p16、p21和γ-H2AX蛋白水平较高,p16、γ-H2AX和53BP1的血管细胞染色丰富。 第二,常氧和慢性缺氧小鼠的肺血管细胞衰老现象。低氧增加了p16LUC/+小鼠的胸腔生物发光。在野生型小鼠中,缺氧增加了肺部的衰老和DNA损伤标志物的水平,衰老相关的分泌表型成分,以及p16对肺内皮细胞(P-ECs,常态下占肺SCs的30%)和肺动脉平滑肌细胞的染色。 第三,消除p16表达细胞加重缺氧诱导的PH和改变正常缺氧小鼠的肺部血流动力学。通过自杀基因或ABT263消除SC增加了右心室收缩压和肥大指数,增加了血管重塑(在常氧和缺氧期间,分裂的增殖细胞核抗原染色的血管细胞数量增加),并明显减少了肺部P-ECs。 第四,药物消除衰老细胞会加剧缺氧和SM22-5-HTT+小鼠的PH,并改变正常缺氧小鼠的肺部血流动力学。为了测试在野生型小鼠中,药物性的衰老细胞消除是否产生与p16-ATTAC小鼠的干预类似的效果,该研究使用了2种衰老药物,ABT263(navitoclax),它通过抵消Bcl2家族成员的抗凋亡功能来诱导衰老细胞的凋亡,以及FOXO4-DRI肽,显示通过损害FOXO4对P53活性的功能来诱导衰老细胞的凋亡。 第五,药物性衰老-细胞消除改变了小鼠暴露于Sugen加Normoxia或Hypoxia的肺部血流动力学特性。由于消除衰老细胞对P-ECs有一贯的有害影响,我们调查了纳维酮(navitoclax)是否会在用Sugen(一种VEGF受体拮抗剂)治疗后改变肺血流动力学。 第六,激活p16或消除p16表达的细胞会改变老龄常氧和低氧小鼠的肺部血流动力学。为了评估p16失活的长期影响,该研究将p16luc/luc小鼠与p16luc/+小鼠以及对照组p16+/+小鼠进行了比较,年龄为14至18个月。与年轻小鼠相比,年长的p16luc/+小鼠的胸腔生物发光增加,在慢性缺氧期间进一步增加。与p16+/+对照组小鼠相比,在常氧和缺氧期间,年长的p16luc/luc和p16luc/+小鼠表现出大量的RVSP增加,肌肉化的肺血管增加,以及通过IB4染色确认的P-ECs的损失。Fulton指数在3个小鼠品系中没有差异,但暴露于慢性缺氧的p16luc/luc小鼠的Fulton指数明显高于其p16luc/+和p16+/+对应的小鼠。 第七,Navitoclax改变了大鼠的肺血流动力学并加重了野百合碱(MCT0)诱导的PH。 通过衰老干预消除衰老的P-ECs可能会使肺部血流动力学恶化。这些结果促使人们考虑在各种情况下旨在控制细胞衰老的策略对肺血管的潜在影响。 研究临床意义 在人类特发性肺动脉高压和实验性肺动脉高压中,衰老细胞在重塑的肺血管和肺毛细血管中积累。使用自杀基因或衰老策略消除衰老细胞改变了健康小鼠和大鼠的肺血流动力学,并加剧了各种损伤诱发的肺动脉高血压。这些对清除衰老细胞的反应的改变主要归因于消除衰老的肺内皮细胞,这导致了大肺血管的重塑和肺毛细血管的损失。 解除衰老的内皮细胞的影响可能会破坏对肺血管细胞生长的控制,从而促进或加剧肺动脉高压的发展或进展。衰老干预在许多情况下被证明是有益的,但在使用时应注意其对内皮细胞和肺血管的潜在影响。

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



