儿童颅内原发Rosai-Dorfman病1例及文献复习
2020-05-26 李汶洋 赵祥 肖格磊 中南大学学报(医学版)

Rosai-Dorfman病(Rosai-Dorfman disease,RDD)又名窦组织细胞增生伴巨大淋巴结病(sinus histiocytosiswith masive lymphadenop
Rosai-Dorfman病(Rosai-Dorfman disease,RDD)又名窦组织细胞增生伴巨大淋巴结病(sinus histiocytosiswith masive lymphadenopathy,SHML),发病率低,是一类原因不明的良性组织细胞增生性疾病,首次报道于1965年,并于1969年被首次确认为一类疾病。而颅内原发性RDD发病率更低,国内外罕有文献报道。因该疾病影像学表现并无明显特征,故初步诊断常与脑膜瘤、胶质瘤或郎汉斯细胞增生症等混淆。本文报告中南大学湘雅医院小儿神经外科收治的第1例儿童颅内原发RDD,并进行相关文献复习。
1. 临床资料
1.1 一般资料
患儿,男,12岁,因发现右额-颞部头皮下肿块2年、头痛、头晕1个月入院。既往史无特殊。体格检查:神志清楚,智力发育正常,右侧额-颞部头皮下可扪及面积约2.5 cm×2.5 cm骨缺损区,相应区域内可扪及一搏动性肿块,质软偏韧,活动度差;双颌下及颈部可触及多个无痛肿大、但活动尚可的淋巴结。
1.2 常规检查
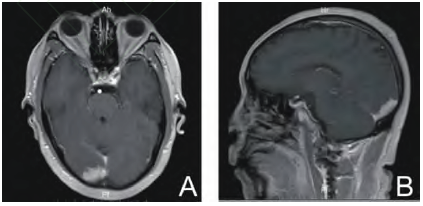
头颅三维CT检查显示:右侧额-颞部一大小约10.4 cm×5.1 cm×8.6 cm的密度较高的肿块,周围有大片水肿带,脑中线左移。骨窗显示:肿瘤对骨质有侵袭性破坏,其余颅骨及颌面骨多发溶骨性破坏(图1A,1B)。磁共振显示肿瘤强化明显,硬脑膜有侵犯,肿瘤“毛刺征”明显,提示软脑膜破坏,与部分脑组织边界不清(图1A)。胸腹部CT显示:双侧肱骨头、双侧肩胛骨、双侧锁骨、胸骨、骶骨、双侧髂骨及多个椎体出现不同程度的溶骨性破坏。单光子发射计算机体层成像术(single-photon emission computed tomography,SPECT)骨扫描显示多处骨代谢增高及降低(图1C)。

图1 患儿影像学资料
实验室检查显示:红细胞沉降率为76 mm/h,白介素-6水平为31.9 pg/mL,均为中度升高。肝肾功能、血尿常规、甲胎蛋白(alpha fetoprotein,AFP)、癌胚抗原(carcinoembryonic antigen,CEA)等未见明显异常。
1.3 手术治疗
入院后为明确诊断,考虑患儿有非手术治疗的可能性,于局麻下行立体定向活检术,取颅骨缺损处皮下2 cm位置小标本送病理检查。病理结果提示:Rosai-Dorfman病,伴较多淋巴细胞、浆细胞浸润,可见组织细胞吞噬淋巴细胞。活检后1个月患儿自觉头痛症状加重,复查磁共振显示肿瘤体积有增大,遂行开颅肿瘤切除术。取右侧额-颞部大问号皮瓣入路,见病变位于额-颞交界处,跨前中颅窝,大小约为10.1 cm×5.2 cm×9.3 cm,质软偏韧,血液运输丰富,相关区域颅骨变薄合并有部分骨质缺损。肿瘤与硬脑膜关系密切,主体位于硬脑膜下,侵及软脑膜,与部分脑组织边界不清,相关区域硬脑膜与软脑膜均不完整。肿瘤予以显微镜下全切除并送病理检查,考虑到病理性质偏良性及患儿美观与功能方面的问题,骨瓣予以保留。
1.4 病理结果
患儿术后最终病理结果(图2)提示右额颞叶Rosai-Dorfman病。免疫组织化学结果如下:白细胞分化抗原163(cluster of differentation 163,CD163)阳性;CD68阴性;CD1a阴性;CD207阴性;CD117阴性;可溶性蛋白-100(soluble protein-100,S-100)阳性;细胞增殖抗原Ki-67约10%阳性;钙黏附蛋白E(E-cadherin)阴性;神经胶质酸性纤维蛋白(glial fibrillary acidicprotein,GFAP)阴性;免疫球蛋白G(immunoglobulin G,IgG)部分阳性;IgG4每高倍镜视野9个阳性细胞;血小板衍生生长因子受体α(platelet-derived growth factor receptor α,PDGFRα)阳性。
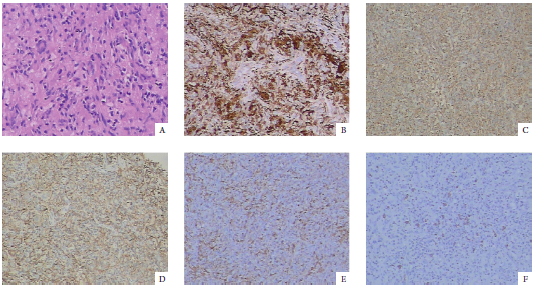
图2 肿瘤切片病理及免疫组织化学结果
1.5 后续治疗
患儿术后出现皮下积液及反复发热等情况,多次抽取积液加压包扎后头皮无法贴附,予以皮下置管引流,1周后引流量减少,患儿皮下积液消失,拔除引流管后未见复发。术后加用激素治疗(泼尼松40 mg口服,每日1次)及化学药物治疗(氨甲喋呤+6-巯基嘌呤+强的松方案联合达沙替尼)。患儿目前化学药物治疗结束,后续病情稳定,随访8个月预后良好,复查未见肿瘤复发迹象(图1D)。
2. 讨论
RDD的典型表现为无痛、对称性双侧颈部淋巴结肿大,伴发热及白细胞升高,常发生于儿童及20岁以下的青少年。但RDD是一类可以发生于全身各系统的良性组织细胞增生性疾病,发生于淋巴系统外的RDD占全部RDD的比例大于40%,故该疾病临床表现形式多样,发生于淋巴系统外的RDD表现出相对应系统的不典型症状。
颅内RDD则经常表现为头痛、呕吐等颅高压症状,以及各种占位效应,如肿瘤累及鞍区可能会造成视力缺损及内分泌功能障碍,累及脑室系统可能会导致梗阻性脑积水,甚至引起昏迷死亡等。
目前,对于颅内RDD的初步筛查与诊断依然缺乏特异性方法,影像学上与颅内脑膜瘤、嗜酸性肉芽肿等疾病容易混淆。目前有部分学者提出了磁共振弥散加权成像(diffusion weighted imaging,DWI)及磁共振表观扩散系数(apparent diffusion coefficient,ADC)来鉴别颅内RDD与脑膜瘤,但受限于敏感性及技术水平等因素,实施起来还是有一定困难。
病理学检查为该病的诊断金标准,RDD在电子显微镜下组织学表现为不同形态的组织细胞、浆细胞与淋巴细胞,形成“明暗相间的结构”。而免疫组织化学染色结果中,S-100与CD68两项结果呈阳性对于RDD的诊断特异性及敏感性均较高。手术治疗为治疗颅内RDD最为直接而有效的方法。
但本例中,因肿瘤体积大,且颅骨骨质破坏较严重,初步考虑为患儿朗格汉斯细胞增生症、血液系统疾病或侵袭性脑膜瘤等的可能性大,存在非手术治疗可能性,且直接手术治疗需要考虑肿瘤切除范围、神经功能保留程度、颅骨是否要丢弃等问题。先行立体定向活体组织检查能够初步帮助判断其病理性质,为进一步治疗提供相关依据。据国内外相关研究报道,除手术治疗外,辅助治疗(放射治疗、化学治疗、激素治疗)对于颅内RDD也可能起到一定的控制与治疗效果,部分病例类固醇治疗效果显著。本例患儿尽管做了肿瘤全切,但其受累的骨质及硬膜范围较广,难以做到全部切除。因此,术后仍有必要进行化学治疗及激素治疗。
目前,有部分学者认为RDD可能是一类IgG免疫相关性疾病,体现在激素治疗对于个体的差异性上。本研究显示:在免疫组织化学结果中,淋巴细胞中IgG4阳性细胞数量占比高的患者类固醇治疗效果要优于IgG4阳性细胞占比低的患者,治疗效果与阳性细胞占比呈正相关关系。这也为激素治疗颅内RDD提供了一定的参考价值。
综上所述,儿童颅内RDD是一类可以采取多学科联合治疗的良性疾病,但该病的治疗目前仍缺少相关规范化的治疗指南,手术治疗是最直接而有效的治疗方法,放射、化学药物和激素治疗可能对部分患儿有效。同时作者认为对于肿瘤累及骨质和硬膜范围较广的患儿,术后也应考虑实施化学药物和激素治疗,以巩固疗效。对于病情较重、肿瘤较大且有进展趋势的患儿(如本病例),明确诊断后以手术为主,联合放射、化学药物和激素治疗等为目前的治疗趋势;手术在考虑保护神经功能的同时力求全切;因手术伤口创面广、瘤腔体积大、硬脑膜破坏严重,须积极有效地预防术后皮下积液及颅内感染等并发症。
原始出处:
李汶洋,赵祥,肖格磊,赵杰,刘景平.儿童颅内原发Rosai-Dorfman病1例及文献复习[J].中南大学学报(医学版),2019(05):600-604.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





学习了
109
#Dorfman#
79
#Rosai-Dorfman病#
99
#OSA#
95
#ROS#
64