JNNP:肌萎缩侧索硬化症系统基因筛查的意义
2021-02-19 MedSci原创 MedSci原创

肌萎缩侧索硬化症(Amyotrophic lateral sclerosis,ALS)是一种以运动皮质、脑干和脊髓上下运动神经元进行性损伤和细胞死亡为特征的成人神经退行性疾病。这导致神经肌肉系统进行性
肌萎缩侧索硬化症(Amyotrophic lateral sclerosis,ALS)是一种以运动皮质、脑干和脊髓上下运动神经元进行性损伤和细胞死亡为特征的成人神经退行性疾病。这导致神经肌肉系统进行性衰竭,大多数病例在症状出现后2-5年内死亡,通常死于呼吸衰竭。高达50%的病例也表现出轻度认知障碍,约5%进展为临床公认的额颞叶痴呆症(FTD)。虽然大多数ALS病例被认为是散发性的(sALS),但5%-10%的ALS病例被证明是家族性的,通常是常染色体显性遗传,约60%-70%的ALS患者是遗传原因家族性肌萎缩侧索硬化症(fALS)的病例已被确定。肌萎缩侧索硬化症最常见的遗传原因是由于C9orf72基因第一内含子中GGGGCC(G4C2)六核苷酸重复序列的扩增。在英国队列研究中,fALS和sALS病例中的这种扩展频率分别为43%和7%,这与全球范围内fALS和sALS的39.3%和7.0%的数据相当。在fALS患者和sALS患者中,还报告了导致ALS的下一个最常见的遗传原因SOD1、TARDBP、和FUS基因突变。
因此,明显的散发病例也可能携带已知ALS基因的潜在致病性变体。在最近一项筛选17个ALS相关基因的研究中,27.8%的明显散发病例携带已知ALS基因中的潜在致病性或罕见变体。目前,至少在英国,只有有ALS家族史、痴呆症或发病年龄较轻的病例才倾向于在临床环境中常规进行基因筛查,随着针对与SOD1或C9orf72突变相关疾病的特定遗传形式的治疗方法的出现,这就提出了是否所有ALS患者都应进行基因筛查的问题。“ALS的多中心生物标志物研究策略”(AMBRoSIA)是一个纵向生物取样计划,在该计划中,新转诊的ALS患者被接触参与研究。同意的患者在研究基础上进行基因筛查,纵向采集血液、尿液和脑脊液样本,同时进行皮肤活检以进行成纤维细胞培养。本文报告了对散发性ALS和家族性ALS患者进行前瞻性基因筛查的结果。
对从英国谢菲尔德运动神经元疾病诊所连续招募的100例ALS患者的前瞻性病例序列中的44个基因组进行了靶向测序。所有受试者均由神经科专家顾问诊断为肌萎缩侧索硬化症。7/100患者有家族性肌萎缩侧索硬化,但多数为散发性肌萎缩侧索硬化。
21%的ALS患者有确诊的或可能的致病突变,其中93%的患者无ALS家族史。15%符合当前ALS基因治疗试验的纳入标准。5/21的致病性突变患者有一个额外的不确定意义的变异(VUS)。另外21%的ALS患者携带ALS相关基因中的VUS。总的来说,13%的患者携带一种以上的遗传变异(致病性或VUS)。携带两种变异体的ALS患者发病年龄明显早于携带单一变异体的患者(发病年龄中位数为56岁vs 60岁,p=0.0074)。
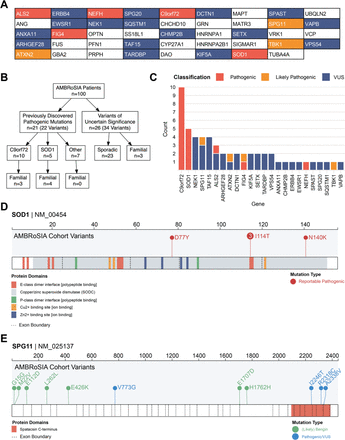
结果表明,筛选已知的ALS基因可以在约21%的患者中产生临床可操作的结果,并且在另外约21%的患者中可以发现具有潜在临床意义的VUS。随着未来研究证实的ALS基因病因数量的增加,这些百分比可能会增加。我们开发了一个利用全基因组病例对照队列来预测临床结果的VUS优先排序管道。虽然这项研究是在一个大型的三级转诊ALS中心进行的,需要在其他环境下进一步验证,本文数据表明,所有ALS患者都应该在仔细的咨询下接受基因检测,特别是考虑到正在开发的新的个性化药物治疗。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#萎缩#
62
#基因筛查#
74
学习了!感谢
102
#硬化症#
100
#肌萎缩#
64
#肌萎缩侧索硬化#
70
学习了,谢谢
109
学习了,谢谢
103