Nature子刊:华人团队通过评估肿瘤总mRNA水平,预测癌症预后并指导用药
2022-06-19 王聪 “生物世界”公众号

对癌症患者肿瘤样本中的肿瘤特异性总 mRNA 水平进行评估,可以作为多种癌症的预后生物标志物。
2022年6月13日,MD 安德森癌症中心 Wang Wenyi 团队在 Nature Biotechnology 期刊发表了题为:Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression 的研究论文。
研究团队开发了一种量化癌症患者肿瘤样本(包括癌细胞和肺癌细胞)中的肿瘤特异性总 mRNA 水平(TmS)的方法,对来自15种不同癌症类型的6580名癌症患者的肿瘤样本进行验证,证实了癌细胞中较高的 mRNA 水平与癌症患者存活率降低有关。
这项研究表明,对癌症患者肿瘤样本中的肿瘤特异性总 mRNA 水平进行评估,可以作为多种癌症的预后生物标志物。

该研究的通讯作者 Wang Wenyi 教授表示,单细胞测序研究显示,癌细胞中的总 mRNA 含量与肿瘤的生物学特征密切相关,但是使用单细胞测序来分析大型患者队形是不可行的。而这项最新研究提出了一种新的数学反卷积技术,通过广泛可用的肿瘤 bulk 测序数据来大规模研究癌症的这一重要生物学特征(癌细胞中总 mRNA 含量)。

Wang Wenyi 教授
单细胞测序技术可以从样本中分析成千上万个单独的细胞,而批量测序可以在大量细胞中生成肿瘤的整体景观。由于从癌症患者中提取的肿瘤样本不仅包含有癌细胞,还包含着非癌细胞,是一个细胞混合物,因此需要额外的步骤来从批量测序数据中分离出癌症特异性测序结果。
反卷积(Deconvolution)是一种计算技术,旨在将批量测序数据分成不同的部分。该研究首次报告了一种从 bulk 测序数据中量化肿瘤特异性总 mRNA 水平的反卷积方法,这为单细胞测序分析提供了可扩展的补充。bulk 测序,提取组织、器官或一群细胞的混合 RNA(bulk RNA)进行测序,得到一群细胞的转录组的平均数据或代表性数据。
为了开发这款反卷积工具,研究团队首先分析了10名患有四种不同癌症类型的患者的48913个细胞产生的单细胞测序数据。将这些测序数据汇集,就像在 bulk 测序样本中一样,使得他们能够识别癌细胞和非癌细胞之间总 mRNA 水平的差异,从而推动进一步检查肿瘤样本 bulk 测序中的这些差异。
在用癌细胞系验证这种反卷积工具后,他们使用来自四个大型队列的15种不同癌症类型的6580个癌症患者肿瘤样本的批量测序数据,量化了其肿瘤特异性总 mRNA 水平。由于 bulk 测序已常规使用多年,研究人员能够将肿瘤特异性总 mRNA 水平与这些患者的长期临床数据进行比较。比较结果显示,较高的肿瘤特异性总 mRNA 水平与更低的无进展生存期和总生存期相关。
此外,该研究还发现,这种相关性还取决于癌症所处的阶段,在有些癌症队列中,特定阶段的癌症高总 mRNA 水平反而与更好的癌症结果相关。对于癌症而言,早期和晚期癌症有不同的治疗方案,研究团队认为总 mRNA 水平可用于预测癌症患者的预后和对某些治疗方法的反应。
研究团队对两个独立的乳腺癌患者队列进行了进一步分析,结果显示较高的总 mRNA 水平与接受化疗的早期阶段乳腺癌患者的预后改善有关,而总 mRNA 水平较低的早期乳腺癌患者似乎从化疗中获益较少。
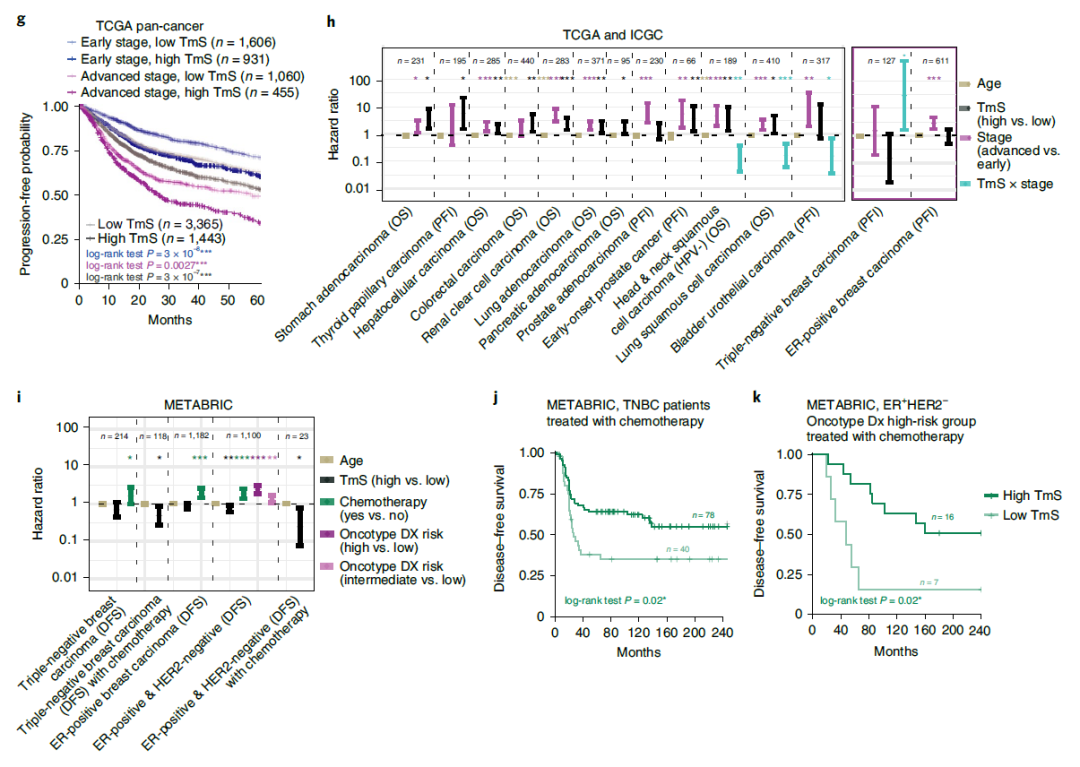
总的来说,该研究证实,高水平的肿瘤特异性总 mRNA 水平(TmS)与癌症进展和死亡风险增加有关。因此,肿瘤特异性总 mRNA 水平(TmS)可以作为癌症的预后生物标志物,对高危癌症患者进行分级并指导治疗选择。
原始出处:
Cao, S., Wang, J.R., Ji, S. et al. Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-022-01342-x.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#Nat#
60
#预测癌症#
65
#华人#
59