
【筛查】拯救“折翼天使”——儿童法布雷病早诊早治
2024-06-03 网络 网络 发表于上海

法布雷病(也称法布里病, Fabry disease),是一种罕见的X染色体伴性遗传溶酶体贮积病。

图源:千库网ID:20506992
(一) 每一个小群体都不该被放弃
2024年4月国家医疗保障局《医保新闻》栏目发布了一则医保动态1,分享了一位来自江西省抚州市的“法布雷”病患者家属的自述:
2021年8月,那是一个平静而悠闲的暑假,孩子们还沉浸在无忧无虑的玩乐当中。一天晚上,我爱人和岳母突然把我叫到卧室,刚进卧室,我爱人就瘫坐在地上开始痛哭:“怎么办啊!我和我妈、还有我弟,我妹妹都确诊了法布雷病,小孩遗传得病的概率很大,医院叫我们带老大老二去抽血寄到上海检查!”那一刻,我仿佛晴天霹雳一般,站在原地久久没回过神。那一晚,我用手机不停地搜索关键词“法布雷”,搜索显示法布雷病是一种罕见的X连锁遗传溶酶体贮积症,会引起多脏器病变甚至引发危及生命的并发症。我心里暗暗一惊,因为“多脏器损伤”“肾衰竭”是小舅子的典型症状,“儿童期四肢剧烈痛”等症状大儿子有过,再往下翻,满屏触目惊心的“年费用上百万”“天价治疗费用”等字眼,更让我整晚辗转反侧,恐惧、无助、绝望在黑夜里弥漫。
行尸走肉般上了半个月班,我还是决定带小孩前往医院抽血,并寄往上海检查。焦急等待一段时间后,基因检查结果出来了,有不幸、有万幸。不幸的是大儿子基因突变了,幸运的是小儿子基因正常。此后,我带着妻儿踏上了漫漫寻医路。在上海瑞金医院办理入院后,母子二人做了全面检查,诊断评估结果显示,大儿子已经开始出现法布雷病的早期症状,得马上用药补充体内缺乏酶,否则将影响脏器功能。但是,医生告诉我们,治疗法布雷的特效药“瑞普佳”单价1.2万一支,还没有进医保目录,需要全额自费,一年治疗费用上百万。那一刻,我们内心本就微弱的希望火苗再次熄灭了。面对高昂医药费用,我们只能抱着孩子无奈打道回府。想起在外就医时,每次给小孩请假,老师都会问,你家小孩怎么总请假,儿子也问,爸爸,我怎么了,为什么总是要打针,而且每次都要抽血,我的血都快抽干了吧?爸爸我会死吗?我只能告诉儿子,没事,你只是身体缺少一种酶,我们补充下就好了。我深怕善意的谎言有一天会被揭穿。
2022年1月,传出天大好消息,我们需要用的法布雷病特效药真的进入了国家医保用药目录,原本需要1.2万一支的瑞普佳,降到了3100元一支!那一刻,压在我心底沉重巨石一下子就落地了。目前,在各级部门的帮助下,孩子得以在当地保障用药,每两周住院治疗,预计全年需要就医26次,总费用预计26万元,医保可报销17.3万元,再加上“抚惠保”等其他保险,预计可再报销5万元,全年个人自付仅约4万元。经过一段时间的治疗,大儿子的病情已经得到了极大改善,四肢疼痛症状缓解了,发热现象也消失了,心肌酶谱恢复正常,肝、肾功能均维持在稳定状态,我们的家庭也恢复了往日的平静。

图源:千库网ID:20613510
(二)儿童期早筛、早诊、早治是关键
法布雷病(也称法布里病, Fabry disease),是一种罕见的X染色体伴性遗传溶酶体贮积病。该病是由于 GLA 基因变异,导致该酶‑α‑半乳糖苷酶 A(α‑galactosidase A, α‑GLA)活性部分或全部丧失,造成其代谢底物三己糖酰基鞘脂醇(globotriasoylceramide, GL‑3)和相关的鞘糖脂在人体各器官、组织的大量贮积,最终引起肾脏、心脏和脑血管等一系列脏器严重损伤。
法布雷病在儿童至青少年期多表现为肢端疼痛。血管角质瘤,易误诊为过敏性紫癜。角膜涡状混浊,需与使用胺碘酮、三苯氧胺、羟氯喹等药物引起者鉴别。还常出现恶心、呕吐,蛋白尿、血尿、肾功能异常。患儿容易怕热但是不出汗。由于临床表现多样,特异性不高,随着 GL 3 的贮积和器官系统受到影响,法布雷病的早期症状进展为危及生命的并发症如慢性肾功能衰竭、心力衰竭、脑卒中时才得以明确诊断。2022年一项大型法布雷病结局调查研究提示,在儿童中延迟诊断的中位年龄为 4~5 年2,3。
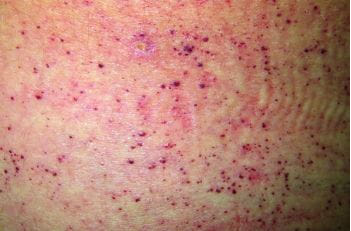
图1法布里病患者的血管角质瘤4
法布雷病早期诊断可提供在不可逆的器官损伤发生之前进行干预的机会。法布雷病的检测方法主要包括α‑Gal A活性检测、基因检测、生物标志物检测如血浆GL‑3、Lyso‑GL‑3水平,以及组织病理学活检等手段。
在治疗方面,国内外指南推荐特异性酶替代疗法(ERT)。ERT通过外源性补充基因重组的α‑Gal A,替代患儿体内酶活性降低或完全缺乏的α‑Gal A,促进GL‑3分解,GL‑3 和 Lyso‑GL‑3在器官组织的贮积减少,阻止或延缓多系统病变发生。ERT治疗可减少症状、改善生活质量、改善临床体征和生化标志物,儿童时期启动ERT治疗可延缓或阻止进展性器官损伤5。
参考资料:
1. 国家医疗保障局(http://www.nhsa.gov.cn/art/2024/4/12/art_14_12357.html)
2. 刘佳璐,徐虹. 中华儿科杂志. 2023,61(09):860-862.
3. Beck M, et al. Orphanet J Rare Dis. 2022,17(1): 238.
4. 默沙东诊疗手册(https://www.msdmanuals.cn/home/children-s-health-issues/hereditary-metabolic-disorders/fabry-disease)
5. Germain DP, et al. Clin Genet. 2019 Aug;96(2):107-117.

如遇到疑似患者,添加微信领取采血包免费安排送检
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#罕见病# #法布雷病#
23