黏多糖贮积症Ⅰ型:症状体征、病因、流行病学、诊断和治疗
2022-08-25 MedSci原创 MedSci原创

黏多糖贮积症是一组由溶酶体异常引起的遗传性黏多糖代谢障碍,系酶的活性缺陷使不完全分解的氨基葡萄糖贮积而引起的先天性风湿病。其中黏多糖贮积症Ⅰ、Ⅳ型最为常见且较具特征性,而尤以Ⅰ型最典型,为黏多糖贮积症
黏多糖贮积症是一组由溶酶体异常引起的遗传性黏多糖代谢障碍,系酶的活性缺陷使不完全分解的氨基葡萄糖贮积而引起的先天性风湿病。其中黏多糖贮积症Ⅰ、Ⅳ型最为常见且较具特征性,而尤以Ⅰ型最典型,为黏多糖贮积症的原型,为常染色体隐性遗传,分为3个亚型:Hurler综合征:即MPS-IH型;Scheie综合征:即MPS-IS型,亦即7大类中原Ⅴ型(MPS-Ⅴ型);Hurler-Sheie综合征:其改变介于前两型之间。
I 型粘多糖贮积症 (MPS I) 是一种罕见的遗传疾病,会影响身体的许多部位(多系统)。根据发病年龄、症状严重程度、疾病进展速度以及是否有早期和直接的大脑受累,患有 MPS I 的儿童被描述为患有严重或减弱(意味着减少)形式的疾病。由于有不同的治疗方案可供选择,因此确定个体是否患有严重或减弱的 MPS I 至关重要。患有严重 MPS I 的个体通常在 6 个月大时出现症状,而患有 MPS I 减毒的个体可能要到 3 岁之后才会变得明显,许多 MPS I 减毒个体直到儿童后期甚至青少年才被诊断出来。患有严重 MPS I 的个体在婴儿早期出现症状,并有早期进行性智力下降的证据,如果未经治疗,会在最初十年内死亡。相比之下,MPS I 减弱的个体出现疾病症状较晚,疾病进展较慢,智力下降,预期寿命接近正常。 MPS I 由 IDUA 基因的变异(AKA 突变或致病序列变异)引起,并以常染色体隐性遗传模式遗传。因此,每个受影响的 MPS I 个体的父母都是 MPS I 的携带者。作为 MPS I 的携带者不会导致症状。
一、一般概述
MPS I 是称为粘多糖贮积症的一组遗传性代谢疾病的成员,而粘多糖贮积症又是称为溶酶体贮积症 (LSD) 的一大类疾病的一部分。溶酶体作为细胞内的主要消化和再循环单位。溶酶体中的酶将特定的细胞成分(如碳水化合物、蛋白质和脂肪)分解或消化成它们的基本单位,然后可以回收利用。健康的细胞和器官不断分解、回收和构建新的细胞成分。在患有 MPS 疾病(包括 MPS I)的个体中,溶酶体酶的缺乏或功能不正常会导致称为糖胺聚糖的特定复合碳水化合物的异常积累。糖胺聚糖曾经被称为粘多糖,这也是这些疾病得名的原因。当细胞不能分解这些糖胺聚糖时,它们就会在各种组织中积累,例如骨骼、关节、大脑、脊髓、心脏、脾脏或肝脏,并导致 MPS I 个体所具有的症状。
最好将 MPS I 视为一系列疾病,范围从生命早期出现的严重形式(Hurler 综合征)到可能直到童年后期才出现的不太严重的形式。患有 MPS I 的个体以前被归类为患有严重、轻度或中间形式的疾病。重度型称为 Hurler 综合征,轻度型称为 Scheie 综合征,中间型称为 Hurler-Scheie 综合征。尽管仍然使用术语 Hurler 综合征,但现在使用术语衰减 MPS I 代替 Hurler-Scheie 和 Scheie。
二、症状与体征
MPS I 中出现的具体体征和症状差异很大,取决于许多因素,包括儿童患有何种形式的 MPS I、何时开始治疗以及个体儿童对各种治疗方案的反应。重要的是要注意 MPS I 是一种进行性疾病,因此儿童的症状也与诊断时儿童的年龄有关。发病年龄、疾病严重程度、智力残疾程度和进展速度在受影响个体之间存在显着差异。
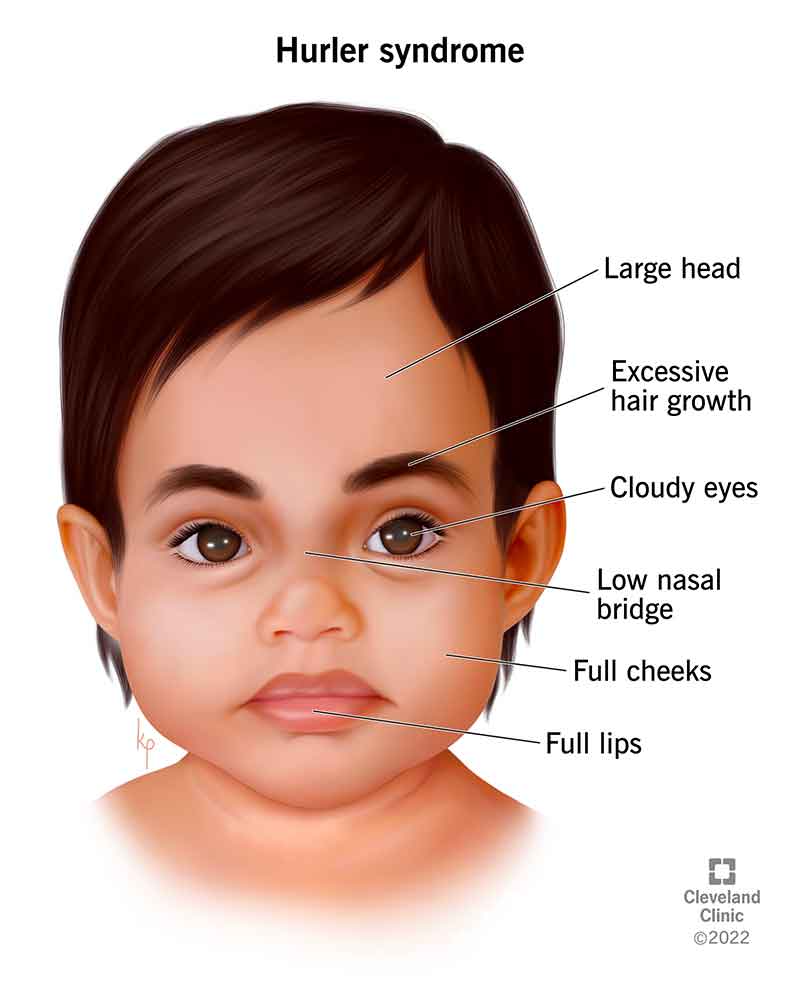
图片:黏多糖贮积症Ⅰ型面容(图源:克利夫兰诊断)
1、重度粘多糖贮积症 I 型
严重形式的 I 型粘多糖贮积症的常见体征和症状是运动技能和智力发育迟缓,以及骨骼畸形。症状可能在出生时不存在,并且可能要到几个月到 1 岁时才会变得明显。
有时,受影响的婴儿有腹股沟或脐疝。腹股沟疝是指一些腹部组织或小肠的一部分穿过腹股沟附近腹部肌肉的隆起或撕裂。脐疝是指一些腹部组织或小肠的一部分穿过肚脐的隆起或撕裂。受影响的婴儿在出生后的第一年经常经历反复的上呼吸道感染。由于疝气和耳部感染在儿童中都很常见,因此仅凭这些发现通常不会导致诊断。
在 1 年至 18 个月大时,受影响的儿童通常
- 发展独特的面部特征,通常被描述为粗糙。这可能包括舌头、嘴唇、鼻孔外侧(鼻翼)和耳垂的增厚。颅骨顶部(颅骨或颅骨)增厚会导致婴儿的头部异常大(大头畸形)。头部最终可能会显得异常长而窄(头颅畸形)。面部和身体的毛发过度生长(多毛症)也可能发生,尽管头皮毛发可能粗而直,类似于茅草或稻草。
- 表现出发育异常的迹象,通常与语言和精细运动技能的获得缓慢有关
- 经常感染中耳(中耳炎),这可能导致听力损失。
- 表现出骨骼疾病的迹象,包括下背部凸出(凸出畸形)和/或关节出现僵硬。
- 角膜混浊(混浊)会随着年龄的增长而恶化,最终导致视力障碍。可能存在称为青光眼的眼内压力增加并导致视力丧失。视网膜可能退化。视网膜是排列在眼睛后部的膜,包含称为视锥细胞和视杆细胞的特殊细胞。这些细胞将光转化为神经冲动,沿着视神经传送到大脑,然后转化为图像。视神经是眼睛的主要神经,也可能发生故障(视神经萎缩)。眼睛异常会导致视力丧失、夜盲症,并最终导致失明。
- 肝脾肿大。医生在检查孩子时会发现这一点,但肝脏和脾脏的肿大本身不会导致症状。
上面列出的迹象代表严重疾病的早期迹象,但由于这种疾病是进行性的,其他潜在症状会随着疾病的进展和儿童年龄的增长而出现,包括:
- 随着金属功能的下降,发育进行性延迟,特别是当治疗没有及早开始时。
- 脑脊液积聚(脑积水)。
- 婴儿和儿童可能会出现发育迟缓或发育不足。身材矮小是指儿童的身高明显低于基于年龄和性别的预期。
- 在所有 MPS I 儿童中均可见进行性骨和关节疾病(称为多发性骨发育不全),并导致
- 所有关节变得僵硬
- 手指僵硬导致“爪手畸形”
- 臀部僵硬
- 膝盖撞击也称为“外翻畸形”
- 脊柱弯曲也称为脊柱侧弯
- 腕管综合征(腕部神经受压)
- 脊椎受压
- 气道或呼吸系统也经常受到影响,并可能导致
- 扁桃体和舌头扩大
- 狭窄的气管
- 由于睡眠时气流受阻,夜间睡眠困难,睡眠呼吸暂停
- 心脏也参与 MPS I,可导致:
- 瓣膜疾病与瓣膜增厚和渗漏最常见的是二尖瓣和主动脉瓣。二尖瓣和主动脉瓣关闭不全是常见的发现。二尖瓣位于心脏的左上腔和左下腔之间;它允许血液从心脏的一个腔室(左心房)流向另一个腔室(左心室)。二尖瓣可能会出现异常增厚,这会使血液倒流(反流),从而导致全身血流减少。主动脉瓣反流是指主动脉瓣变厚并允许血液从主动脉(身体的主要动脉)回流到左心室。主动脉瓣关闭不全会导致呼吸急促、胸痛和潜在的快速心力衰竭。其他心脏并发症可能包括心律不齐(心律失常)、左上心室(左心室)过度生长(肥大)和冠状动脉疾病。
- 心脏也可以直接受累,也称为心肌病。
- 眼睛或涉及并导致
- 角膜混浊
- 眼压升高(青光眼)
- 视网膜疾病:眼睛后部受累
- 听力损失在 MPS I 中也很常见
2、减轻型粘多糖贮积症I型
被描述为患有 MPS I 减毒形式的儿童在受严重影响的患者中经历了类似的体征和症状,但往往表现出疾病进展较慢和症状发作年龄较晚。重症患者和减毒患者之间的一个关键区别在于,减毒患者不会表现出早期发育迟缓,也不会经历心智能力的进行性下降。作为一个群体,减毒患者的症状和进展速度表现出显着差异。一些儿童受到轻微影响,寿命接近正常,而另一些儿童则在儿童时期出现症状,通常在 6 或 7 岁左右,并可能在青少年时期或 20 多岁时出现危及生命的并发症。减轻形式的这些症状如上文针对严重形式所述。症状可能与严重形式一样显着,也可能较轻。 MPS I 减弱的个体的生活质量会受到显着影响。
MPS I 减弱儿童的智力通常不受影响,但一些儿童和年轻人可能会出现学习障碍。受影响的儿童会有不同程度的生长不足。他们在青少年时期可能会出现肝脏扩大(肝肿大)、角膜混浊和心脏瓣膜异常。角膜混浊会导致严重的视力问题。也可能发生其他眼部异常,包括青光眼、视神经萎缩和视网膜退化。进行性心脏瓣膜问题可以在 10 或 11 岁时开始。也可发生冠心病。
多发性骨发育不全、骨骼畸形、腕管综合征和进行性关节疾病也可能以减毒形式发生。这些体征和症状与上述严重形式的相同。受影响的人也可能有一个高足弓(pes cavus)和弯曲的膝盖,以便在腿伸直时它们相互摩擦(膝盖或膝外翻)。有些孩子可能会用脚掌走路,这样脚后跟就不会接触地面(脚趾走路)。
中度至重度听力损失也可能以衰减形式出现。有些孩子可能会出现睡眠呼吸暂停。疝气也可能存在。脊髓的进行性压迫也是可能的,并且可能导致包括运动不耐受和活动减少在内的问题。
三、病因
I 型粘多糖贮积症是由 α-L-艾杜糖醛酸酶 (IDUA) 基因的变异(突变、致病序列变异)引起的。基因为创造在身体的许多功能中起关键作用的蛋白质提供了指导。当基因发生突变时,蛋白质产物可能有缺陷、效率低下、缺失或过度生产。根据特定蛋白质的功能,这会影响身体的许多器官系统,包括大脑。
IDUA 基因调节α-L-艾杜糖苷酶的产生。需要这种酶来分解体内产生的称为糖胺聚糖(过去称为粘多糖)的复杂碳水化合物。当 IDUA 基因发生改变时,功能性 α-L-艾杜糖醛酸酶水平不足。如果没有适当水平的这种酶,这些糖胺聚糖,尤其是硫酸皮肤素和硫酸乙酰肝素,就会在所有细胞的溶酶体中积聚。这种异常积累会干扰细胞的正常功能和健康,最终导致组织进行性损伤并导致症状。在严重的 MPS I 中,完全没有 α-L-艾杜糖苷酶,而在减毒形式中,细胞中可能产生非常非常少量的酶。
遗传疾病是由我们从父母那里获得的基因组合决定的。我们都有 2 个 IDUA 基因拷贝;我们从父亲那里继承了一份,从母亲那里继承了一份。当一个人有 2 个基因拷贝并且两个拷贝都存在错误时,就会以隐性方式遗传疾病。所有 MPS I 个人都是如此;他们有 2 个被改变的 IDUA 基因拷贝;他们从各自的父母那里收到了一份。因此,父母是 MPS I 的携带者。携带者具有 IDUA 基因的一个正常拷贝和一个改变拷贝。携带者是健康的,作为 MPS 的携带者,我不会导致症状。两个携带者父母都通过改变的 IDUA 基因并因此生育受影响的孩子的风险是每次怀孕的 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。孩子从父母双方那里获得正常基因的机会是 25%。男性和女性的风险相同。
四、流行病学
粘多糖贮积症 I 型影响男性和女性的数量相等,重度型的发病率约为 1/100,000 活产儿,而减轻型的发病率约为 500,000 名活产儿中的 1 例。 发病率是在给定时间段(例如一年)内发生疾病的人数。 MPS 疾病的发病率总计约为 25,000 分之一。 然而,罕见的疾病,尤其是较轻的 MPS,经常被误诊或漏诊,因此很难确定普通人群中的真实频率。
五、鉴别诊断
以下疾病的症状可能与 I 型粘多糖贮积症相似。比较可能有助于鉴别诊断。
溶酶体贮积病是遗传性代谢疾病,其特征是由于酶缺乏导致身体细胞中各种物质的异常积聚。 这些疾病总共有近 50 种,它们可能影响身体的不同部位,包括骨骼、大脑、皮肤、心脏和中枢神经系统。 不断发现新的溶酶体贮积症。 虽然对其中一些疾病的可能治疗方法正在进行临床试验,但目前许多溶酶体贮积病尚无批准的治疗方法。
六、诊断
粘多糖贮积症 I 型的诊断基于特征性症状的识别、详细的患者和家族史、全面的临床评估和各种专业测试。有特征性早期体征的婴儿可能会怀疑诊断。目前,MPS I 的新生儿筛查正在许多地区实施。
临床测试和检查
对 MPS I 患者的尿液进行专门检查可以发现糖胺聚糖(粘多糖)水平升高,特别是乙酰肝素和硫酸皮肤素。这不是 MPS I 的诊断,但表明粘多糖贮积症。
MPS I 的明确诊断需要检测特定细胞,例如白细胞(白细胞)或结缔组织细胞(成纤维细胞)。这些测试证明了 α-L-艾杜糖苷酶的低活性。
分子遗传学检测通常用于确认诊断。分子遗传学检测可以检测已知会导致 MPS I 的 IDUA 基因中的致病改变(突变),但只能在专业实验室作为诊断服务使用。
在美国,MPS I 的新生儿筛查最近被批准纳入推荐的通用筛查小组,尽管每个州独立决定何时或是否将其添加到其新生儿筛查小组中。新生儿 MPS I 筛查的一个问题是,在某些情况下,医生可能无法区分患有 MPS I 的新生儿是否会发展为严重或轻度的疾病。由于这些形式的治疗方法不同,这可能会导致确定适当治疗过程的挑战。目前正在进行研究,以寻找区分患有严重 MPS I 的婴儿与患有轻度 MPS I 的婴儿的方法。
七、治疗
MPS I 的治疗包括三个组成部分:
1)更换缺失的酶
2) 减轻疾病的特定症状
3) 为家庭提供遗传咨询
1) 替代缺乏酶:溶酶体酶是一种独特的蛋白质,因为它们可以被细胞吸收利用。因此,治疗 MPS I 的一种潜在方法是为患者提供他们缺少的 α-L-艾杜糖苷酶。这可以通过两种方式完成。一种方法是给它们注入纯化的酶,也称为酶替代疗法 (ERT)。酶替代疗法包括用基因工程(重组)形式替代缺失的酶,α-L-艾杜糖苷酶。这是通过每周通过静脉将酶注入患者体内来进行的。 2003 年,美国食品药品监督管理局 (FDA) 批准 laronidase (Aldurazyme) 用于治疗大多数 MPS I 患者。Aldurazyme 对大脑和中枢神经系统损伤相关的症状没有帮助,因为药物不能通过血液-脑屏障,它是血管和细胞的保护网络,允许一些物质进入大脑,同时阻止其他物质进入。
另一种替代患者体内酶的方法是进行造血干细胞 (HSCT) 或骨髓移植。造血干细胞是在骨髓中发现的特化细胞(在长骨中发现的柔软的海绵状物质)。这些造血干细胞生长并最终发育成三种主要类型的血细胞之一——红细胞、白细胞或血小板。进行移植以用没有 MPS I 的人的骨髓替换受影响个体的骨髓。移植产生的健康细胞含有足够水平的白细胞,这些白细胞会产生 α-L-艾杜糖苷酶,然后这些细胞代替组织中的酶缺乏症。该手术费用高昂,并存在严重并发症的风险,包括死亡、移植物抗宿主病和其他长期和晚期影响。 HSCT 被认为是严重 MPS I 的护理标准,因为它是目前唯一可用于改善严重 MPS I 个体智力成果的治疗方法。
尽管 HSCT 和酶替代疗法可以显着改善临床过程,但它们并不能治愈这种疾病,因此,随着年龄的增长,许多受影响的个体仍然面临重大挑战和问题。一般来说,越早开始治疗效果越好,疾病的严重程度和儿童的年龄通常会影响手术的结果。 HSCT 对智力障碍的影响可能会有所不同。通常,一旦出现严重的智力障碍,就无法逆转,尽管认知能力下降可以得到缓解。详细见:NEJM:自体造血干细胞基因疗法治疗黏多糖贮积症I型患儿
2) 缓解疾病的特定症状:I 型粘多糖贮积症治疗的一个重要组成部分是针对每个人明显的特定症状。这需要一个专家团队的协调努力,包括在内。
儿科医生
- 心脏病专家:专门诊断和治疗心脏病的医生
- 神经科医生:专门诊断和治疗神经系统疾病的医生
- 骨外科医生、风湿病学家、物理治疗师、职业治疗师:专门诊断和治疗骨骼和关节疾病
- 耳鼻喉科医生 (ENT):治疗扁桃体肿大、上呼吸道和听力损失
- 眼科医生:治疗眼睛和视觉症状
- 呼吸科医师:气道疾病的治疗
3) 建议对受影响的个人及其家人进行遗传咨询。对整个家庭的社会心理支持也很重要。
资源部分中列出的几个组织提供有关 MPS 疾病(如 MPS I 型)的支持和信息。
八、预后
本型为进行性,4岁开始严重衰退,常在10岁前死亡。死亡年龄较其他类型黏多糖贮积症早,死因多为心功能不全。Hurler综合征患者常于儿童期死亡,Scheie综合征及Hurler-Scheie综合征可存活至成年。
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Kuiper GA, Langereis EJ, Breyer S, et al. Treatment of thoracolumbar kyphosis in patients with mucopolysaccharidosis type I: results of an international consensus procedure. Orphanet J Rare Dis. 2019;14:17. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6339313/
Donati MA, Pasquini E, Spada M, Polo G, Burlina A. Newborn screening in mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):126. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6238254/
Grosse SD, Lam WKK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet Med. 2017;19:957-982. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5763496/
Parini R, Deodato F, Di Rocco M, et al. Open issues in mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12:112. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5472858/
Khan SA, Peracha H, Ballhausen D, et al. Molecular genetics and metabolism. Mol Genet Metab. 2017;121:227-240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5653283/
Peake RWA, Bodamer OA. Newborn screening for lysosomal storage disorders. J Pediatr Genet. 2017;6:51-60. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5288002/
Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12:32. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5307824/
Laraway S, Mercer J, Jameson E, et al. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J Pediatr. 2016;178:219-226. https://www.sciencedirect.com/science/article/pii/S0022347616307004
Kunin-Batson AS, Shapiro EG, Rudser KD, et al. Long-term cognitive and functional outcomes in children with mucopolysaccharidosis (MPS)-IH (Hurler syndrome) treated with hematopoietic cell transplantation. JIMD Rep. 2016;29:95-102. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5059216/
Khalid O, Vera MU, Gordts PL, et al. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis type I. PLoS One. 2016;11:e0150850. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4795702/
Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164-2172. http://www.bloodjournal.org/content/125/13/2164
Wraith JE, Jones S. Mucopolysaccharidosis type I. Pediatr Endocrinol Rev. 2014;12 Sup 1:102-106. https://www.ncbi.nlm.nih.gov/pubmed/25345091
Wilkinson FL, Holley RJ, Langford-Smith KJ, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7:e35787. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3338781/
Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183-1197. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3228957/
De Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3170181/
Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology (Oxford). 2011;50 Suppl 5:v19-25. https://academic.oup.com/rheumatology/article/50/suppl_5/v19/1780256
Muenzer J, Wraith JE, Clarke LA, et al. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19-29. https://www.ncbi.nlm.nih.gov/pubmed/19117856
Clarke LA. Mucopolysaccharidosis Type I. 2002 Oct 31 [Updated 2016 Feb 11]. In: Pagon RA, Bird TD, Dolan CR, et al., GeneReviews. Internet. Seattle, WA: University of Washington, Seattle; 1993-. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1162/ Accessed February 18, 2019.
Genetic and Rare Diseases Information Center. Mucopolysaccharidosis Type I. December 2012. Available at: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i Accessed February 15, 2019.
Beck M. Mucopolysaccharidosis Type 1. Orphanet Encyclopedia, October 2011. Available at: https://www.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=EN&Expert=579 Accessed February 15, 2019.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#流行病#
65
认真学习~~
70