

儿童横纹肌肉瘤(rhabdomyosarcoma)是发自间叶组织的恶性肿瘤,占儿童实体肿瘤的15%,软组织肉瘤的50%。根据数据显示,该病在0至14岁儿童癌症病例中的发病率为2.7%,在15至19岁青少年和年轻人中为1.4%。每年约有350例新病例发生,其中50%出现在前10岁。
从1975年至2020年,儿童和青少年癌症患者的生存期得到持续改善,儿童癌症死亡率下降了50%以上。1975年至2017年间,15岁以下儿童横纹肌肉瘤患者的5年相对生存率从53%增加到71%,15至19岁青少年患者的5年相对生存率从30%增加到52%。
2020年,世界卫生组织将横纹肌肉瘤分为四种组织学亚型:胚胎型、腺泡型、梭形细胞/硬化型和多形性。其中,FOXO1基因融合仅发生在腺泡型组织学亚型中。男性患者胚胎型肿瘤发病率较高,而黑人患者腺泡型肿瘤发病率略高。
世界卫生组织(WHO)第5版软组织和骨骼肿瘤分类中确认四种组织学类别具有独特的基因组改变。
胚胎型横纹肌肉瘤其特点是11p15位点杂合度缺失,RAS通路基因突变频率高。与腺泡型横纹肌肉瘤相比,胚胎型横纹肌肉瘤患者被认为PAX3-FOXO1和PAX7-FOXO1基因融合阴性(即融合阴性横纹肌肉瘤)。
腺泡横纹肌肉瘤以FOXO1与PAX3或PAX7基因融合为特征(即FOXO1融合阳性横纹肌肉瘤)。组织学上没有FOXO1基因融合的腺泡型横纹肌肉瘤与胚胎型横纹肌肉瘤有相似的临床行为、基因改变模式和转录组谱。
梭形细胞/硬化性横纹肌肉瘤的特点是老年患者存在MYOD1突变,幼儿患者存在VGLL2和NCOA2基因重排。
多形性横纹肌肉瘤以复杂的核型为特征,具有数量上的和不平衡的结构变化,与未分化多形性肉瘤难以区分。
在缺乏PAX-FOXO1基因融合(融合阴性横纹肌肉瘤)和PAX-FOXO1基因融合(融合阳性横纹肌肉瘤)的患者中,基因突变和基因扩增(CDK4和MYCN)的分布是不同的。
以下是融合阴性(FN)和融合阳性(FP)横纹肌肉瘤患者基因改变的频率
Nras 17%/ 1%,KRAS9%/ 1%,Hras 8% /2%,Fgfr4 13%/ 0%,
Nf1 15%/ 4%,Bcor 15% /6%,Tp53 13%/ 4%,Ctnnb1 6% /0%,
Cdk4 0% /13%,Mycn 0% /10%。
融合阴性横纹肌肉瘤(胚胎组织学) 胚胎型横纹肌肉瘤通常表现为11p15位点杂合性缺失,8号染色体上杂合性增加。与腺泡横纹肌肉瘤相比,胚胎型肿瘤具有更高的背景突变率和更高的单核苷酸变异率,且随着诊断年龄的增长,体细胞突变数量增加。最常见的复发突变包括RAS途径中的突变(例如,NRAS, KRAS, HRAS和NF1),大约有一半的病例观察到这些突变。NRAS突变是婴儿期以后最常见的RAS途径基因突变,而HRAS突变主要发生在婴儿期。目前看RAS突变的存在并不具有预后意义。
在RAS通路基因中,NF1和HRAS的胚系突变易导致横纹肌肉瘤。在一项对615名横纹肌肉瘤儿童的研究中,347名儿童的肿瘤具有胚胎组织学特征。其中,9例患者有NF1胚系突变,5例患者有HRAS胚系突变,分别占胚胎组织学病例的2.6%和1.4%
融合阴性横纹肌肉瘤中出现重复突变的其他基因包括FGFR4、PIK3CA、CTNNB1、FBXW7和BCOR,所有这些基因都出现在不到15%的病例中。
TP53突变 在融合阴性横纹肌肉瘤患者中观察到10%-15%的患者携带TP53突变,而在腺泡型横纹肌肉瘤患者中较不常见(约4%)。在其他儿童癌症(如肾母细胞瘤)中,TP53突变与间变组织学相关,胚胎横纹肌肉瘤也是如此。在一项对146名已知TP53状态的横纹肌肉瘤患者的研究中,大约三分之二的TP53突变肿瘤表现为无间变(69%),但只有四分之一的无间变肿瘤有TP53突变。
在COG和英国横纹肌肉瘤队列的非风险层和风险层分析中,TP53突变的存在与EFS(无事件生存)降低相关。胚胎型和腺泡型患者均观察到与TP53突变相关的不良预后。基于这些结果,COG计划在其即将进行的试验中将TP53突变视为一种高风险的决定性特征。
横纹肌肉瘤是一种与Li-Fraumeni综合征相关的儿童癌症。在一项对614名横纹肌肉瘤儿童患者的研究中,11名患者(1.7%)有TP53生殖系突变。与非腺泡组织的患者(2.2%)相比,有腺泡组织的患者突变较少(0.6%)。具有非腺泡间变性形态的横纹肌肉瘤可能是患有Li-Fraumeni综合征和胚系TP53突变的儿童的表现特征。
DICER1突变 在一小部分胚胎型横纹肌肉瘤患者中观察到DICER1突变,大多数宫颈胚胎型横纹肌肉瘤均有DICER1突变。相比之下,在阴道原发部位的患者中很少观察到DICER1突变,这种情况主要发生在2岁或3岁以下的女孩身上。DICER1突变在子宫体发生的胚胎性横纹肌肉瘤中也很常见,但这种表现主要见于成人。颈部横纹肌肉瘤通常表现为葡萄样肉瘤的组织学模式,许多病例表现为软骨分化区,这一特征也见于其他携带DICER1突变的肿瘤类型。为了支持具有DICER1突变的胚胎型横纹肌肉瘤的独特生物学,这些病例具有与其他胚胎型横纹肌肉瘤不同的DNA甲基化模式。颈横纹肌肉瘤的诊断是DICER1综合征基因检测的指征
融合阳性横纹肌肉瘤(腺泡组织学) 约70%至80%的腺泡型肿瘤以13号染色体上的FOXO1基因与2号染色体上的PAX3基因(t(2;13)(q35;q14)或1号染色体上的PAX7基因(t(1;13)(p36;q14)之间的移位为特征。其他罕见的融合包括PAX3-NCOA1和PAX3-INO80D。PAX3基因易位发生在大约60%的腺泡横纹肌肉瘤病例中,而PAX7基因发生的比例约为20%。具有实体腺泡组织学的患者PAX-FOXO1基因融合的发生率低于典型腺泡组织学的患者。无论是否有转移,与PAX7基因相关的腺泡组织学似乎发生在更年轻的年龄。PAX3基因重排相比PAX7基因重排,具有更短的EFS率。具有腺泡组织学和PAX3基因的患者年龄较大,侵袭性肿瘤(T2)发生率较高。约20%的病例显示腺泡组织学未检测到PAX基因易位。融合阴性腺泡横纹肌肉瘤的临床行为、基因改变模式和转录组谱与胚胎型横纹肌肉瘤患者一致,现在与胚胎型横纹肌肉瘤一起被归为融合阴性横纹肌肉瘤。
除了FOXO1重排外,与融合阴性肿瘤相比,腺泡肿瘤的TMB更低,重复突变的基因更少。融合阳性肿瘤中最常观察到的改变是CDK4的局灶性扩增(13%)或MYCN(10%),少数患者有其他基因的重复突变(例如,BCOR,6%;NF1, 4%;TP53, 4%;PIK3CA, 2%)。腺泡型横纹肌肉瘤中TP53突变似乎意味着治疗失败的高风险。
梭形细胞/硬化性组织学 在WHO软组织和骨骼肿瘤分类中,梭形细胞/硬化性横纹肌肉瘤被作为一个单独的实体提出在梭形细胞/硬化性横纹肌肉瘤类别中,一些实体具有独特的分子和临床特征。
先天性/婴儿梭形细胞横纹肌肉瘤 一些报道描述了涉及VGLL2和NCOA2基因融合的先天性或婴儿梭形细胞横纹肌肉瘤病例(例如,VGLL2-CITED2, TEAD1-NCOA2, VGLL2-NCOA2, SRF-NCOA2)。
对于先天性/婴儿梭形细胞横纹肌肉瘤,一项研究报道11例患者中有10例出现复发性融合基因。这些患者大多为截断型原发肿瘤,没有睾丸旁肿瘤。7例患者(63%)观察到新的VGLL2重排,包括4例患者的VGLL2-CITED2融合和2例患者的VGLL2-NCOA2融合,三名患者(27%)存在不同的NCOA2基因融合,包括两名患者的TEAD1-NCOA2和一名患者的SRF-NCOA2。在本报告中,所有具有长期随访数据的融合阳性先天性/婴儿梭形细胞横纹肌肉瘤患者均存活良好,无患者发生远处转移
虽然大多数先天性/婴儿梭形细胞横纹肌肉瘤的研究都显示出良好的预后,但据报道,4名患者发展为转移性疾病,2名患者结局致命。疾病进展发生在诊断后的中位3.5年(范围1-8年)。所有四名患者都患有无法切除的肿瘤,并接受了化疗。然而,大多数文献报道的病例都是手术切除的。在疾病进展中,一名患者的肿瘤出现TP53突变,另一名患者的肿瘤出现纯合子CDKN2A和CDKN2B缺失。
一项对40例先天性/婴儿梭形细胞横纹肌肉瘤(诊断年龄≤12个月)患者的研究发现,几乎所有患者都有局限性疾病(n = 39),一半接受分子检测的患者(26例中13例)有NCOA2和/或VGLL2重排。由于检测仅限于NCOA2和VGLL2,更全面的基因组分析可能会识别出更高比例的相关基因融合患者。13例VGLL2和/或NCOA2融合患者的5年EFS率为90%,总生存率为100%。
VGLL2、NCOA2和其他相关基因基因重排在儿童先天性/婴儿梭形细胞横纹肌肉瘤中的患病率和预后意义并不明确 。
MYOD1突变型梭形细胞/硬化性横纹肌肉瘤 在年龄较大的儿童和成人梭形细胞/硬化性横纹肌肉瘤中,在很大比例的患者中观察到特定的MYOD1突变(p.L122R)。在COG和英国横纹肌肉瘤患者的联合队列(n = 641)中,在所有融合阴性横纹肌肉瘤病例中,有3%(17/515例中的17例)发现MYOD1突变,而在融合阳性病例中未发现MYOD1突变。MYOD1突变患者的发病年龄为10.8岁。该队列中的大多数病例表现为纺锤形或硬化特征,但也观察到类似胚胎横纹肌肉瘤致密模式的细胞密集排列的病例。该队列中的大多数病例(17例中的15例,88%)原发部位为头颈部或脑膜旁区。在大约一半的MYOD1突变病例中可以看到PIK3CA突变的激活。MYOD1突变的存在与局部和远端失败的风险显著增加相关。
骨内梭形细胞横纹肌肉瘤 原发性骨内横纹肌肉瘤是一种非常罕见的横纹肌肉瘤。大多数病例出现TFCP2基因重排,FUS或EWSR1基因重排。伴有FUS-TFCP2或EWSR1-TFCP2基因融合的横纹肌肉瘤最常见于年轻人,但也有报道在年龄较大的儿童和青少年中发生病例。颅面骨是最常见的原发肿瘤部位,免疫组化ALK和细胞角蛋白阳性是常见的。该实体的其他特征包括复杂的基因组谱,大多数病例显示CDKN2A肿瘤抑制基因缺失伴有FUS-TFCP2或EWSR1-TFCP2基因融合的骨内梭形细胞横纹肌肉瘤具有侵袭性临床过程。在一项研究中,中位OS仅为8个月。
不同的基因突变不同的亚型其预后是不相同的(参见下图)
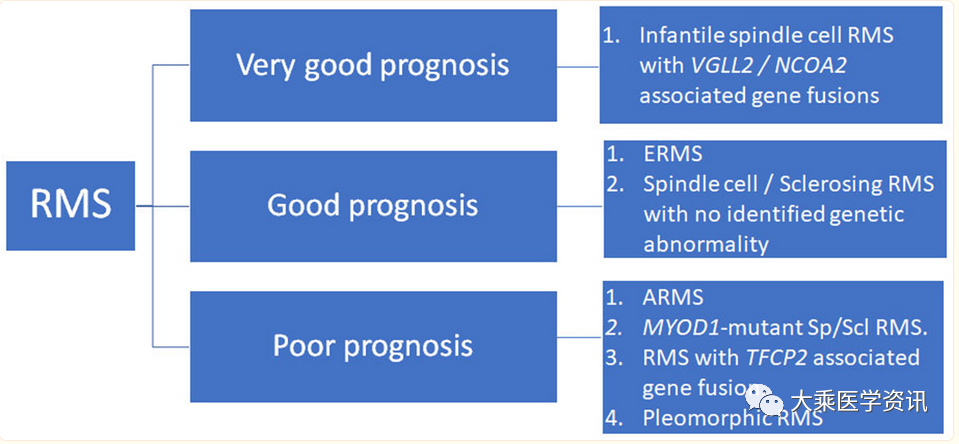
横纹肌肉瘤的分子遗传学目前是研究的热点,分子研究发现基因异常可能导致信号转导、染色质修饰、细胞周期等方面的改变,从而促进肿瘤的发生和发展。对于这些基因异常的深入研究,有助于发现横纹肌肉瘤的新靶点和治疗策略。未来,希望能够通过更深入的分子遗传学研究和治疗方案的优化,为儿童横纹肌肉瘤的治疗带来更好的效果。
与肿瘤大于5厘米的儿童相比,肿瘤小于5厘米的儿童生存率提高。肿瘤体积和最大肿瘤直径均与预后相关。[证据级别C1]
一项对儿童和青少年软组织肉瘤的回顾性研究表明,用于成人软组织肉瘤的5厘米截距对于较小的儿童,尤其是婴儿可能并不理想。该综述确定了肿瘤直径与BSA之间的相互作用这在一项针对中危横纹肌肉瘤患者的COG研究中未得到证实这种关系需要前瞻性研究来确定观察的治疗意义。
Resectability
初次手术后的疾病程度(即手术病理组,也称为临床组)与预后相关。在IRS-III研究中,初次手术后出现局部、大体残留病变的患者(外科-病理组III)的5年生存率约为70%,相比之下,手术后无残留肿瘤的患者(I组)的5年生存率超过90%,手术后有微观残留肿瘤的患者(II组)的5年生存率约为80%。I组和II组代表少数患者;大约50%的患者在诊断时患有不可切除的III组疾病
无功能损害的可切除性与肿瘤的初始大小和部位有关,与疾病的生物学不相关。使用多模式治疗可优化预后。所有患者都需要化疗,至少85%的患者也受益于放射治疗,即使对不可切除疾病的患者也有良好的结果。在IRS-IV研究中,III组局部不可切除疾病患者接受化疗和放疗,5年FFS率约为75%,局部控制率为87%。两项中危COG横纹肌肉瘤研究(D9803和ARST0531 [NCT00354835])合并评估延迟一期切除的益处。在D9803研究中,部分或完全切除后的局部对照放疗在第12周完成。在ARST0531研究中,放射治疗在第4周开始。膀胱或前列腺横纹肌肉瘤患者接受延迟一期切除术后的生存率无差异,而四肢横纹肌肉瘤或非膀胱/非前列腺非四肢横纹肌肉瘤患者接受延迟一期切除术后OS改善。降低辐射剂量的延迟初次切除策略可使这些部位获得更好的OS。
组织病理学亚型
儿童横纹肌肉瘤的肺泡亚型在临床特征较差的患者中更为常见(例如,小于1岁或大于10岁,四肢和躯干原发肿瘤,诊断时转移性疾病)。与胚胎型横纹肌肉瘤的类似患者相比,它通常与较差的结局相关。
在IRS-I和IRS-II研究中,即使在原发肿瘤完全切除的患者中,肺泡亚型也与较差的结果相关(I组)
对1258例IRS-III和IRS-IV型横纹肌肉瘤患者进行分析,发现其5年生存率存在统计学意义上的差异(胚胎型横纹肌肉瘤为82%,肺泡型横纹肌肉瘤为65%)
在IRS-III研究中,I组肺泡亚型肿瘤患者的预后与其他I组肿瘤患者相似,但肺泡患者接受了更强化的治疗。
有局部淋巴结受累的肺泡横纹肌肉瘤患者的预后明显差于没有局部淋巴结受累的患者(5年FFS率,43% vs. 73%)。
肺泡肿瘤和胚胎肿瘤患者放射治疗后的局部控制率相似。然而,出现5厘米或更大肿瘤的患者局部失败率明显更高。
在13%的胚胎横纹肌肉瘤病例中观察到无间变,一些研究表明存在无间变对中危性疾病患者的临床结局有不利影响。然而,间变尚未被证明是一个独立的预后变量。
PAX3:FOXO1或PAX7::FOXO1基因融合状态
大约80%的横纹肌肉瘤在形态学上被定义为肺泡型横纹肌肉瘤表达FOXO1融合。FOXO1基因融合仅发生在肺泡组织学肿瘤中。一些回顾性研究发现融合状态是一个独立的预后因素。易位阴性的肺泡型横纹肌肉瘤患者的肿瘤基因和分子特征与胚胎型横纹肌肉瘤患者相似
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




儿童横纹肌肉瘤
61
不错,学习了。
59