
Circulation 张岩-董尔丹/张抒扬-胡晓敏/李铤团队合作揭示细胞外RIPK3加剧心脏缺血/再灌损伤的作用机制
2024-11-28 论道心血管 论道心血管

北大等团队发现细胞外 RIPK3 与 PCI 术后 MACE 正相关,其作为 DAMP 经 RAGE/CaMKII 通路加重心脏 I/R 损伤,为相关防治提供新靶点与视角。
心肌缺血是由于冠状动脉斑块破裂或侵蚀等导致的冠状动脉血流受阻的病生理过程,是引起心肌损伤的最主要的原因之一。治疗心肌缺血的最佳方法是通过经皮冠状动脉介入治疗(percutaneous coronary intervention, PCI)、冠状动脉旁路移植术(coronary artery bypass grafting, CABG)或溶栓治疗等,及时、充分地恢复冠状动脉血供(即再灌)。然而,再灌过程本身也会引起不可逆的心脏损伤和心肌细胞死亡,称为心肌缺血/再灌(ischemia /reperfusion, I/R)损伤。目前临床对心脏缺血/再灌损伤缺乏有效的干预手段,使其成为当前缺血性心脏病防治的难点。同时,虽然PCI是目前治疗心脏缺血损伤的重要手段,但是超过30-40%的患者在PCI术后五年内会发生主要不良心血管事件(major adverse cardiovascular events, MACEs)。提前对病人MACE的发生风险进行分层,进而对不同风险的病人精准干预,对提高MACE的防治效果具有重要意义。但目前尚无有效的临床标志物来识别残余风险和高风险患者。
受体相互作用蛋白激酶3 (receptor interact protein kinase 3, RIPK3)在调节炎症信号和细胞死亡途径中发挥重要作用。在细胞内,RIPK3与受体相互作用蛋白激酶1 (RIPK1)相互作用形成坏死小体,激活混合谱系激酶结构域样蛋白(MLKL)并介导细胞程序性坏死。张岩研究员的前期工作证实RIPK3通过激活钙/钙调蛋白激酶II (Ca2+/calmodulin protein kinase II, CaMKII),在缺血和氧化应激引发的心肌细胞程序性坏死中发挥着至关重要的作用,抑制RIPK3可减轻包括心脏I/R损伤和心力衰竭在内的多种心脏疾病(Nat Med. 2016, 22,175)。张抒扬/胡晓敏课题组前期研究表明,RIPK3可在外周血中被检测到,能够作为多种心血管疾病的生物标志物(Chin Med J (Engl). 2019,132,1400; Clin Chim Acta. 2020,509, 273)。但是,细胞外RIPK3是否具有生物学功能尚不清楚。
2024年11月22日,北京大学基础医学院心血管研究所/北京大学第三医院张岩研究员团队和董尔丹院士团队,联合北京协和医院心内科张抒扬教授和胡晓敏副研究员团队以及西安交通大学第一附属医院李铤副研究员团队在缺血性心脏病和心力衰竭治疗方面取得重要进展,在美国心脏协会(AHA)主办的国际权威期刊Circulation杂志(中科院一区,2024年影响因子35.5)发表了题为“Extracellular RIPK3 acts as a damage-associated molecular pattern to exaggerate cardiac ischemia /reperfusion injury”的研究论文。美国阿尔伯特-爱因斯坦医学院Wilf Family心血管研究所主任、调控性心肌细胞死亡研究先驱Richard N. Kitsis教授为本文撰写评述,题为“Extracellular role for the intracellular cell death mediator RIPK3 in myocardial infarction”。该研究发现了细胞外RIPK3蛋白在心脏I/R损伤的诊断与防治中的新作用。这项研究首先证实急性心肌梗死(acute myocardial infarction, AMI)患者的血浆RIPK3浓度与MACE的发生呈正相关,是PCI术后MACE风险分层的重要生物标记物。同时揭示了RIPK3作为损伤相关分子模式(damage associated molecular pattern, DAMP)在细胞外的新功能,还发现了其通过与糖基化终末产物受体(receptor of advanced glycosylation end-products, RAGE)结合并激活CaMKII信号通路,从而加剧心脏I/R损伤。这一发现为心梗患者PCI术后的MACE事件风险分层与防治提供了新的视角,同时阐明了外源性RIPK3通过RAGE/CaMKII信号通路在心脏I/R损伤中扮演着致病角色,为心肌I/R损伤及其并发症的治疗提供了一个有潜力的新靶点。

首先,研究者分别招募了接受PCI治疗的AMI患者,建立发现队列(103例,北京协和医院)和验证队列(334例,西安交通大学第一附属医院),通过5年的随访,记录MACE预后信息。在发现队列中,AMI患者入院时的血浆RIPK3水平显著高于健康对照志愿者,患者PCI术后6小时血浆RIPK3水平相比术前进一步升高,且24小时仍维持升高状态。在PCI术后3个月和5年中发生MACE的患者PCI术后6小时血浆RIPK3浓度明显高于无MACE患者。Kaplan-Meier分析显示,血浆RIPK3水平较高的患者3个月和5年MACE风险增加。在验证队列中,研究者同样发现短期随访和长期随访的PCI术后患者MACE组血浆RIPK3水平均明显高于无MACE组,进一步验证了AMI接受PCI治疗患者的细胞外RIPK3浓度与MACE呈正相关。
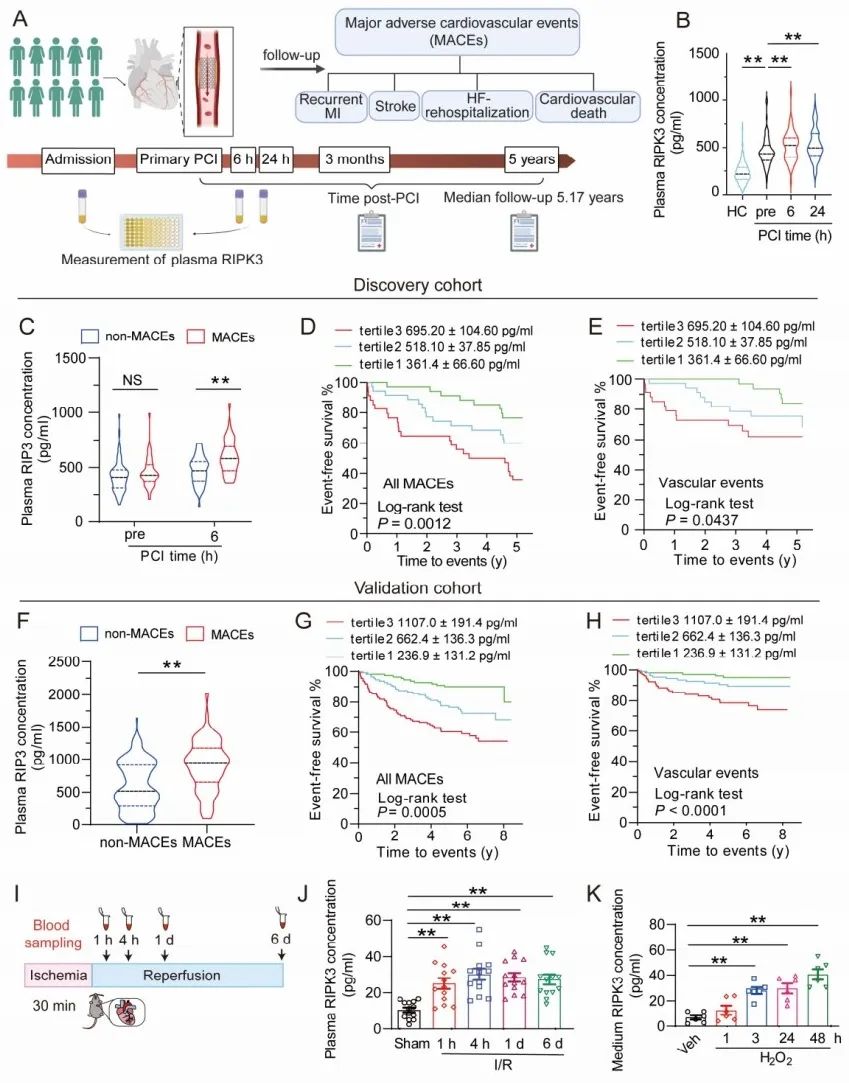
图1. 接受PCI治疗的AMI患者细胞外RIPK3浓度与MACE风险正相关
为探索细胞外RIPK3的来源,研究者采用I/R手术的小鼠模型及缺氧/复氧(H/R)处理的乳大鼠心肌细胞(NRVMs)、炎症细胞、成纤维细胞和内皮细胞模拟PCI引起的心脏I/R损伤过程,发现造模后小鼠血浆和心肌细胞培养上清中RIPK3浓度显著升高,由此可见心脏I/R损伤后血浆升高的RIPK3主要来源于心肌细胞。
接下来,研究者尝试探索细胞外RIPK3在心脏I/R损伤中的病理作用。在I/R条件下,术后连续5天给予小鼠rh-RIPK3 (RIPK3重组蛋白),造成I/R诱导的心肌梗死面积显著增加,且心功能恶化、心肌细胞死亡增多。相反,利用RIPK3中和抗体(anti-RIPK3)阻断细胞外RIPK3,小鼠心肌梗死面积与IgG对照组处理的小鼠相比显著减少。此外,阻断细胞外RIPK3可减轻心功能障碍,并减少心肌细胞死亡。以上证据表明,细胞外RIPK3对心肌I/R损伤是充分必要的。

图2. 细胞外RIPK3加重了小鼠心脏I/R损伤,而中和抗体发挥治疗效应
为了进一步研究细胞外RIPK3诱导的心脏损伤的细胞机制,研究者采用H/R刺激NRVMs、LPS刺激炎症细胞模拟体内心脏I/R损伤,发现给予rh-RIPK3显著增加了心肌细胞死亡和炎症,并上调巨噬细胞的炎症水平。同时在I/R损伤小鼠心脏组织和分离出的外周血单个核细胞(peripheral blood mononuclear cells, PBMCs)分别观测到细胞死亡和炎症加剧,心脏组织巨噬细胞浸润增加,而拮抗细胞外RIPK3则部分缓解这些表型。这些结果表明,细胞外RIPK3加重I/R诱导的心肌细胞死亡和炎症反应。
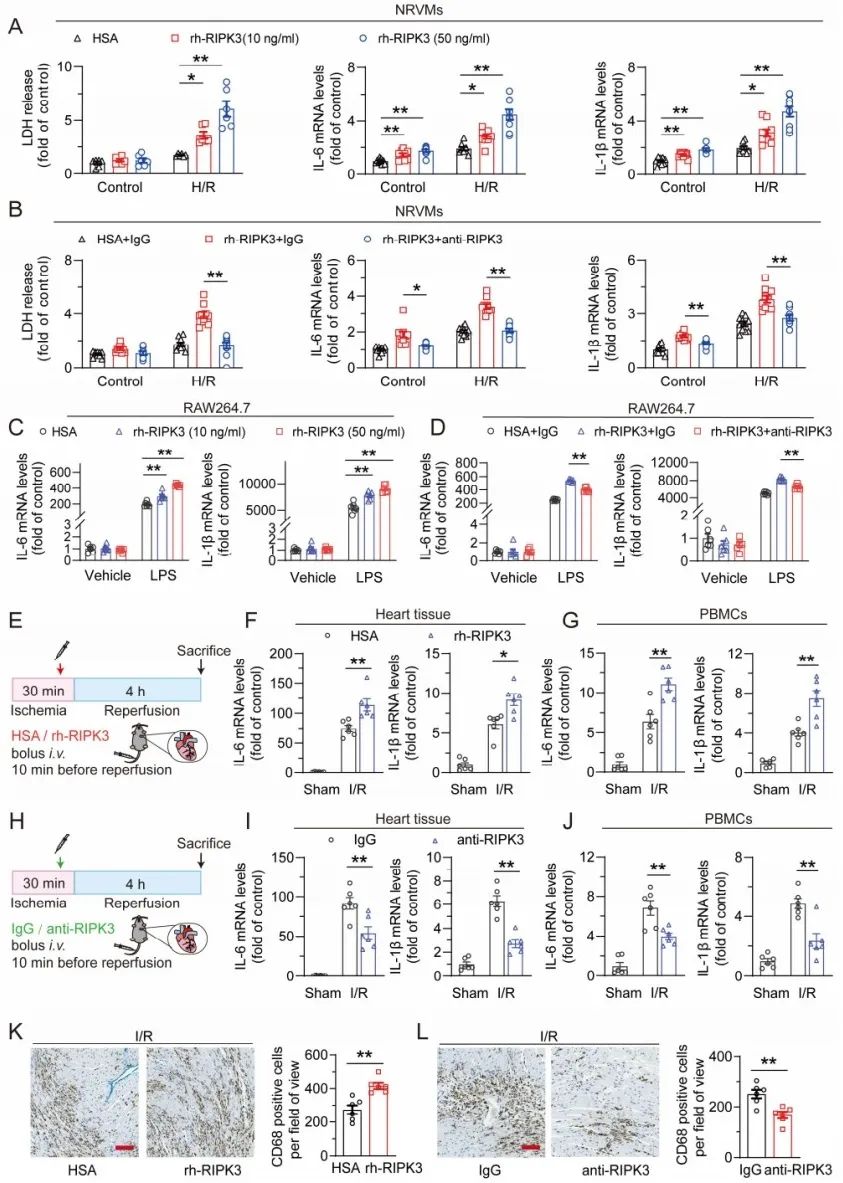
图3. 细胞外RIPK3加重了离体与在体模型中心脏I/R诱导的细胞死亡与炎症
在I/R过程中,炎症和氧化应激引起内皮细胞炎症和凋亡,进而引起内皮损伤和微血管功能障碍。因此,研究者接下来研究了细胞外RIPK3对内皮细胞和相关血管稳态的影响。研究者利用H/R损伤的人主动脉内皮细胞(HAECs)为模型,发现H/R损伤后内皮功能障碍(以MCP-1、VCAM和ET-1的表达为指标)和细胞死亡(以培养基上清中LDH浓度为指标)显著增加,rh-RIPK3处理后进一步加重,而anti-RIPK3处理后得到减轻。为了探索细胞外RIPK3在内皮依赖性血管舒张中的作用,研究者从大网膜中分离了人源动脉,离体动脉分别用HSA或rh-RIPK3孵育8小时,发现rh-RIPK3培养的动脉内皮依赖性血管舒张受到损害。因此,在培养细胞和离体动脉中,细胞外RIPK3均可引起内皮细胞损伤和功能障碍。以上结果表明,细胞外RIPK3通过促进心肌细胞死亡、巨噬细胞炎症和内皮细胞功能障碍,引起心肌微环境紊乱,介导心脏I/R损伤。
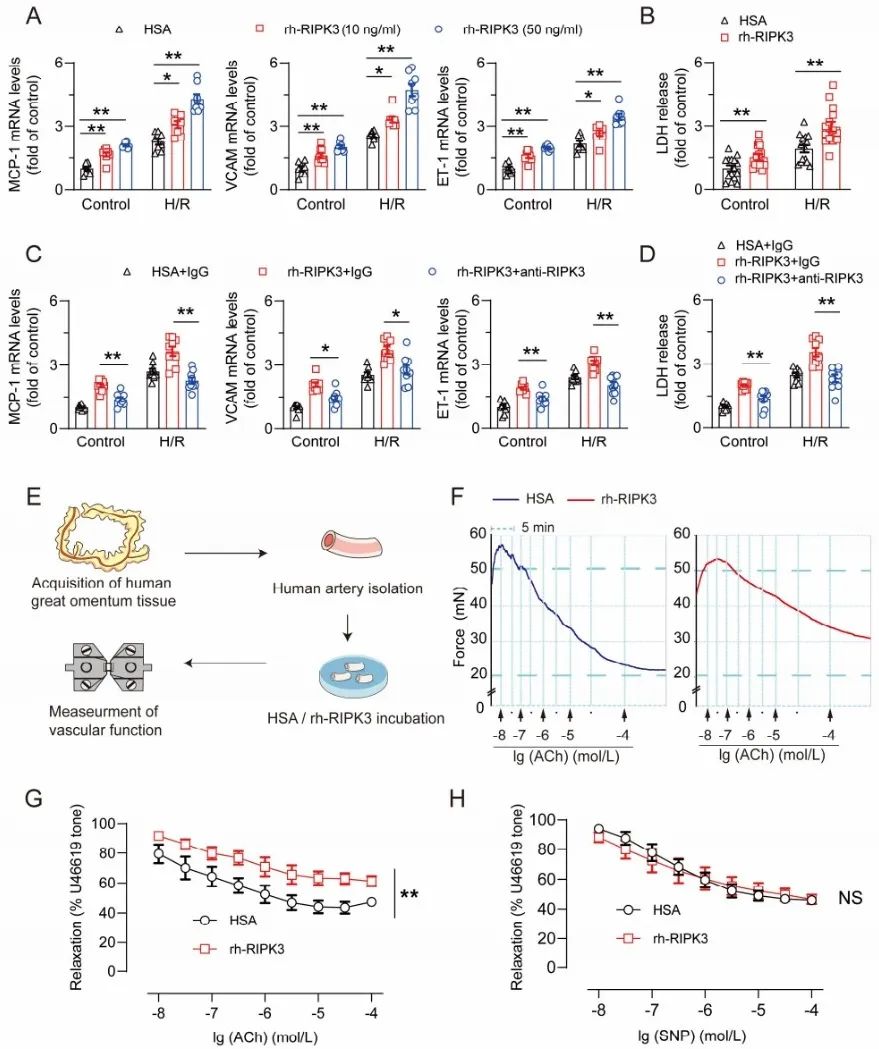
图4. 细胞外RIPK3引起内皮细胞损伤和功能紊乱
接下来,研究者尝试探索细胞外RIPK3介导的细胞死亡和炎症的分子机制。在I/R损伤中,DAMPs的释放是细胞损伤或应激的结果,可激活模式识别受体(PRRs)并导致额外的细胞损伤和炎症。由于以上结果显示RIPK3具有与DAMP高度类似的生物学效应,研究者推测RIPK3发挥DAMP的作用,并通过与PRRs的相互作用诱导炎症和细胞死亡。通过文献调研,研究者对广泛表达、具有蛋白类配体,且先前报道与心血管疾病相关的TLR2、TLR4、RAGE、TLR9和TREM1进行筛选,以确定RIPK3的受体。筛选发现RAGE敲低抑制rh-RIPK3诱导的心肌细胞炎症,且表面等离子共振实验证实RIPK3与RAGE直接结合。据报道,RAGE参与多种细胞的炎症和损伤。因此,研究者进一步探索了RAGE在细胞外RIPK3诱导的病生理效应中的作用。在心肌细胞中,RAGE敲低明显减轻rh-RIPK3诱导的心肌细胞炎症和死亡的加重。RAGE敲低抑制rh-RIPK3孵育后炎症细胞中IL-1β和IL-6的表达上调,并改善rh-RIPK3诱导的内皮损伤和细胞死亡。RAGE受体基因敲除(AgerKO)小鼠相较野生型小鼠,在rh-RIPK3诱导下具有较轻的心肌损伤和炎症。以上数据表明,RAGE是细胞外RIPK3的受体,在RIPK3介导的细胞损伤中发挥重要作用。
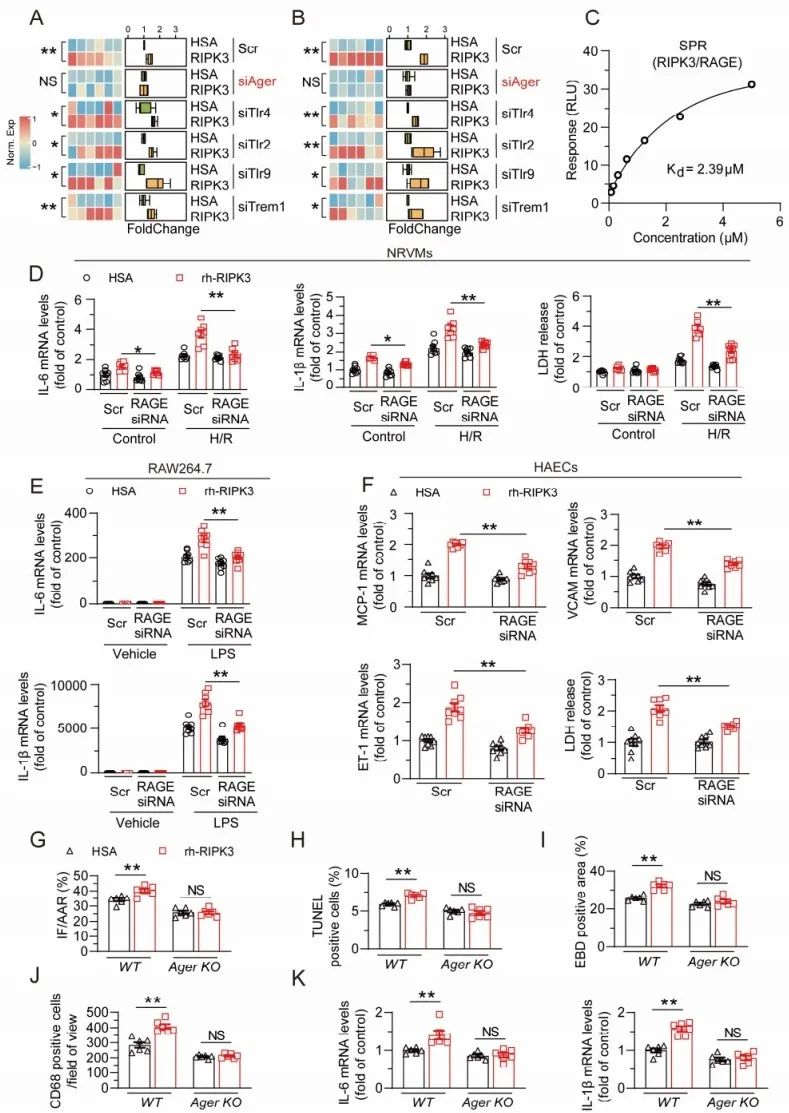
图5. 细胞外RIPK3与RAGE直接结合,并以RAGE依赖的方式介导心脏I/R损伤
张岩研究员的前期工作证实在心肌细胞中,CaMKII是RAGE的重要下游分子,介导细胞损伤和炎症作用(JCI Insight. 2016;1:e84969)。因此研究者提出CaMKII可能介导了RIPK3/RAGE下游的病理效应。发现注射rh-RIPK3可显著提高小鼠心脏I/R后缺血心脏组织和PBMC中CaMKII的磷酸化水平,而拮抗RIPK3得到缓解。在H/R损伤的NRVMs中结果一致。此外,CaMKII抑制剂鲁索利替尼(ruxolitinib)有效抑制rh-RIPK3孵育心肌细胞和巨噬细胞的死亡和炎症效应。在体内条件下,接受I/R手术的Camk2d敲除小鼠中,rh-RIPK3处理未能加重心肌细胞死亡、巨噬细胞浸润和促炎因子的表达。此外,在心肌细胞、内皮细胞和巨噬细胞中,RAGE的敲低可缓解rh-RIPK3诱导的CaMKII过度激活。这些发现表明,细胞外RIPK3与RAGE相互作用,通过激活CaMKII介导进一步的细胞损伤。
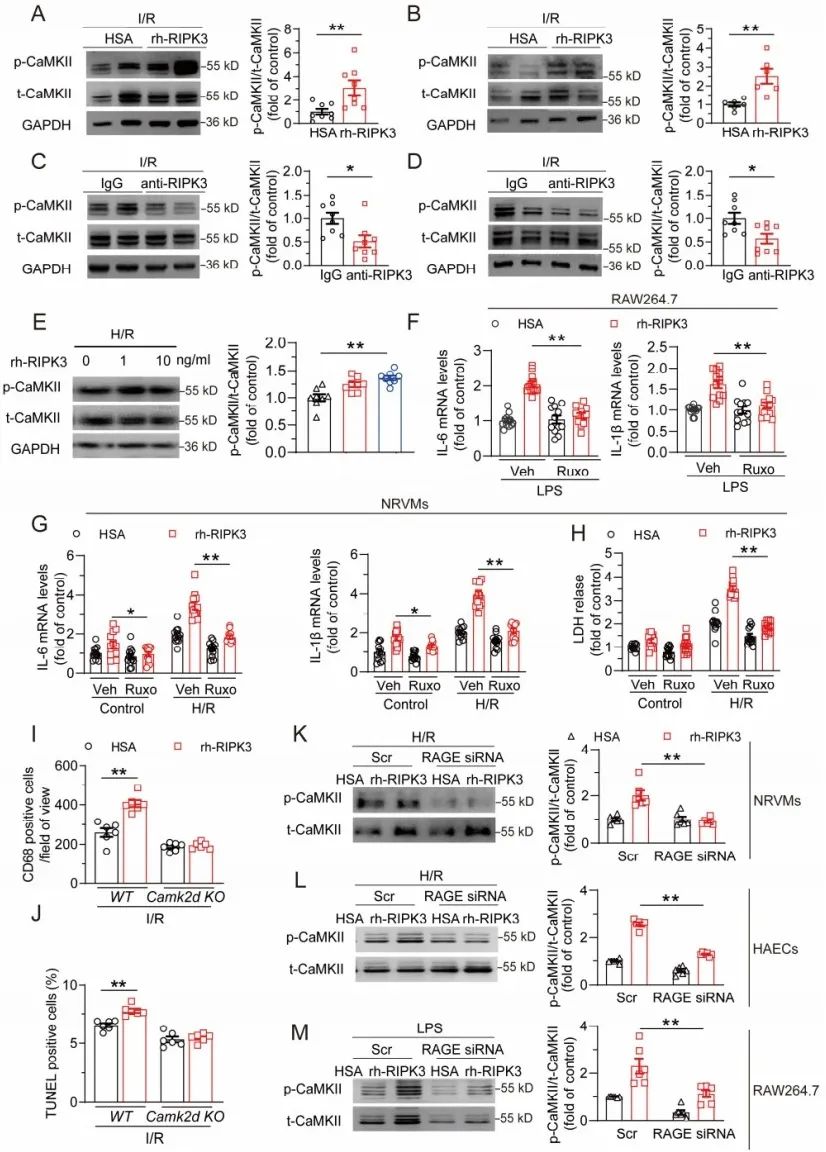
图6. 细胞外RIPK3-RAGE通过激活CaMKII引起损伤效应
最后,为进一步验证上述研究的临床相关性,研究者从PCI术后6小时采集的患者血液样本中分离出人类PBMCs,并与健康对照组同时检测CaMKII磷酸化水平。PCI术后患者血浆RIPK3浓度及PBMCs中CaMKII的磷酸化水平相比健康对照组均明显升高。最重要的是,CaMKII磷酸化水平与血浆RIPK3浓度呈正相关(r = 0.55, P < 0.01),表明细胞外RIPK3浓度与细胞内CaMKII的磷酸化之间存在关联。这些数据有力地支持了PBMCs中CaMKII磷酸化水平的增加可能归因于心脏中的细胞外RIPK3。
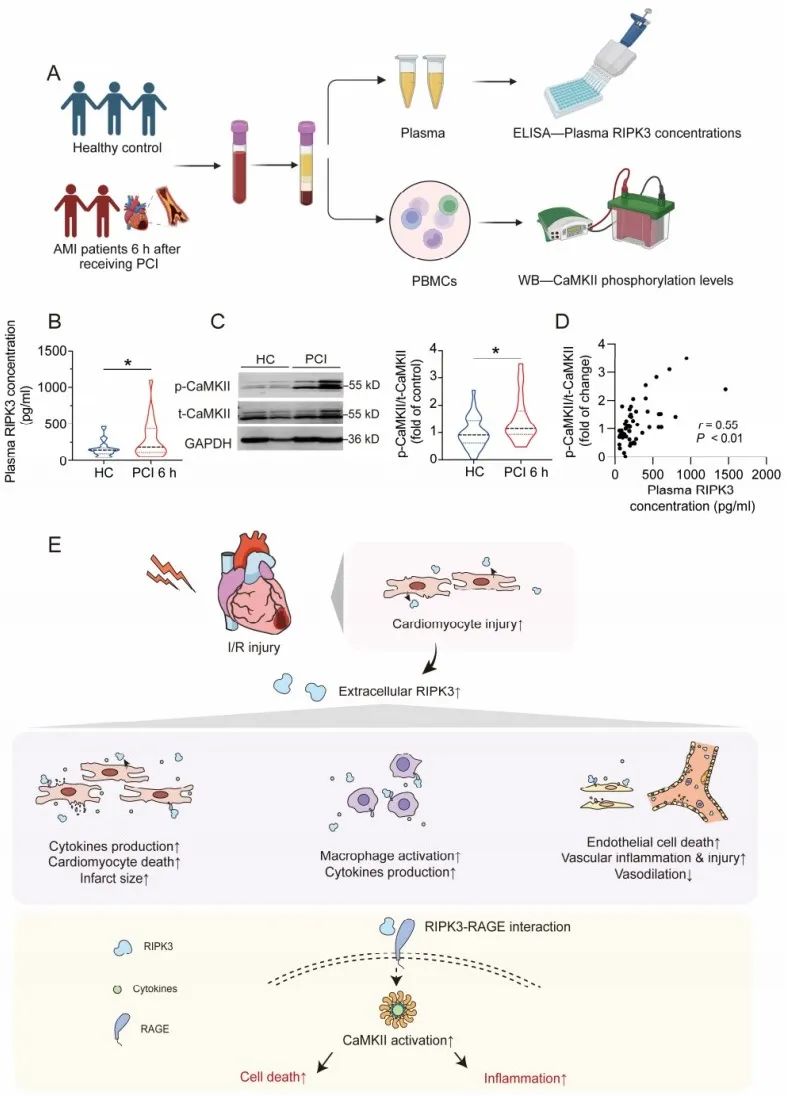
图7. 细胞外RIPK3作为DAMP,通过RAGE-CaMKII通路加重心脏缺血/再灌注损伤机制图:I/R损伤中,受损心肌细胞释放RIPK3。细胞外RIPK3作用于心肌细胞、炎症细胞和内皮细胞,引起损伤、炎症和功能紊乱。机制上,RIPK3可与RAGE互作,促进CaMKII磷酸化活化,进而介导细胞死亡和炎症
综上,接受PCI手术的AMI患者的血浆RIPK3浓度与MACE的发生呈正相关,是PCI术后MACE风险分层的重要生物标记物。除作为生物标记物外,细胞外RIPK3还作为新型DAMP分子,参与介导心肌I/R损伤。这项研究阐明了细胞外RIPK3通过RAGE/CaMKII信号通路在心脏I/R损伤中发挥促炎作用,为心肌I/R损伤及其并发症的治疗提供了一个有潜力的新靶点,同时为PCI术后缺血性心脏病患者的MACE事件风险分层与防治提供了新的视角,对提高MACE的防治效果具有重要意义。
北京大学医学部基础八年制毕业生张文佳、北医三院心内科博士后张俊霞、北京协和医院心内科博士生王泽源和西安交通大学附属第一医院心内科李铤教授为文章的第一作者。此外,本工作获得了北京大学刘雅涵副研究员、汪锴副研究员、王世强教授、郭宇轩研究员,北京大学第三医院彭颖副主任医师、李楠研究员,北京协和医院田庄教授、刘震宇教授、金晔副研究员、张越伦副研究员、郭帆主治医师,西安交通大学附属第一医院吴岳教授、卓小桢副研究员,中国中医科学院石晓路研究员,浙江大学方向明教授和北京安贞医院王媛研究员等的大力支持与帮助。
论文链接:
https://www.ahajournals.org/doi/abs/10.1161/CIRCULATIONAHA.123.068595
述评链接:
https://www.ahajournals.org/doi/full/10.1161/CIRCULATIONAHA.124.072172
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#RIPK3# #心脏缺血/再灌损伤#
16