
J Clin Invest 中山大学附属第一医院黄展鹏团队揭示CARDINAL通过调控翻译改善心肌肥厚
2024-05-15 论道心血管 论道心血管 发表于上海

本研究通过系统性的筛选发现了CARDINAL是心肌特异的核糖体结合蛋白,阐明了CARDINAL通过降低DRG1蛋白稳定性而抑制翻译,从而改善心肌肥厚的作用和其具体分子机制。
引言
心肌肥厚是心力衰竭发生发展过程中的重要病理生理过程。核糖体的翻译功能增强是心肌肥厚的重要特征。也是导致心脏重量增加的重要原因。已有研究表明,抑制核糖体的翻译功能可以有效改善心肌肥厚,提示靶向核糖体功能很可能是心衰治疗的有效策略。然而,已有的研究多关注调控核糖体的蛋白,这些蛋白多数在体内各种组织中普遍表达。考虑到核糖体翻译功能是细胞的重要生物学过程,系统性地抑制翻译很可能导致心脏外器官的不良反应。例如雷帕霉素虽然能有效抑制心肌肥厚,但是也可以导致免疫抑制等严重后果。这些不良反应显然对于心力衰竭治疗时不能接受的。那么,如何才能特异地抑制心肌的翻译呢?
近年来多个研究表明,很多长链非编码RNA(long non-coding RNA, lncRNA)都可与核糖体结合。有趣的是,这些结合核糖体的lncRNA大多并不编码蛋白。这些lncRNA与核糖体结合的生物学意义目前仍不明确,但其中一个合理的猜想是:这些lncRNA可以通过结合核糖体调控核糖体的翻译功能。考虑到部分lncRNA具有很强的组织特异性,若能找到心肌特异的核糖体结合lncRNA,它们很有可能是特异调控心肌翻译的因子,成为未来治疗心力衰竭的有效靶点。
中山大学附属第一医院黄展鹏研究员与哈佛大学波士顿儿童医院Dazhi Wang教授团队(现任南佛罗里达大学Morsani医学院再生医学中心主任)、中山大学附属第一医院心内科何建桂教授团队合作,就上述科学问题进行了系统性的筛查并发现了CARDINAL这一心肌特异的核糖体结合lncRNA,进一步深入探讨了CARDINAL对心肌翻译和心肌肥厚的调控作用和分子机制。相关工作近日以题为“The long non-coding RNA CARDINAL attenuates cardiac hypertrophy by modulating protein translation”的研究论文在线发表在中科院一区的医学权威杂志Journal of Clinical Investigation (IF=15.9)上。

结果
1. 系统性筛查发现CARDINAL是心肌特异的核糖体结合lncRNA
研究先从心肌特异性入手,在人和小鼠的多组织RNA测序数据库中,筛选心脏表达明显高于其他组织的lncRNA。此步筛选发现了5条候选lncRNA。对人体外诱导分化的心肌细胞的多聚核糖体组分和无核糖体组分测序结果分析表明,在5条lncRNA中,CARDINAL与核糖体的结合强度最大。所以锚定CARDINAL进行进一步的研究。
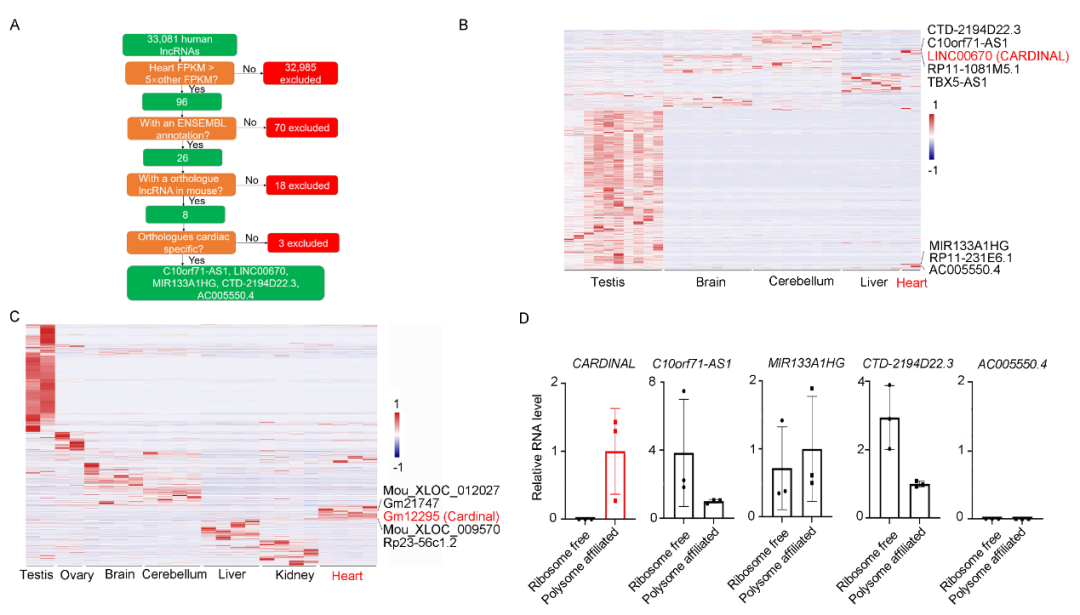
进一步分析发现,在心肌肥厚发生时,核糖体结合的CARDINAL含量升高,提示两者结合可能与心肌肥厚相关。为了进一步验证CARDINAL与核糖体结合,将HL-1心肌细胞的裂解液进行密度梯度离心并分离出不同的组分进行RNA提取和qPCR分析,结果发现,核糖体亚基、单体、多聚核糖体上的CARDINAL显著高于无核糖体组分中的CARDINAL。
在验证了CARDINAL与核糖体的结合后,进一步验证CARDINAL的心肌特异性。提取小鼠不同组织的RNA进行qPCR分析,证实了心脏中的CARDINAL显著高于其他组织(见原文补充材料)。进一步分析CARDINAL具体在心脏中的哪种细胞中高表达,通过Langendroff的方法分离了小鼠心脏的心肌和非心肌细胞,发现CARDINAL主要表达于心脏中的心肌细胞。
完成以上验证后,下一步开始对CARDINAL的一些基础特性进行探究。RACE实验和Northern Blot提示CARDINAL是一长约3kb的lncRNA。Ribo-seq的结果提示CARDINAL的前两个外显子很可能介导了CARDINAL与核糖体结合,但PhyloCSF编码性评分并不支持该区域能编码微肽,提示CARDINAL的确是一lncRNA。RNA-FISH提示CARDINAL在细胞核和细胞浆都有分布,定量分析提示约60% CARDINAL分布与细胞浆。核质分离和qPCR分析结果与此相一致。
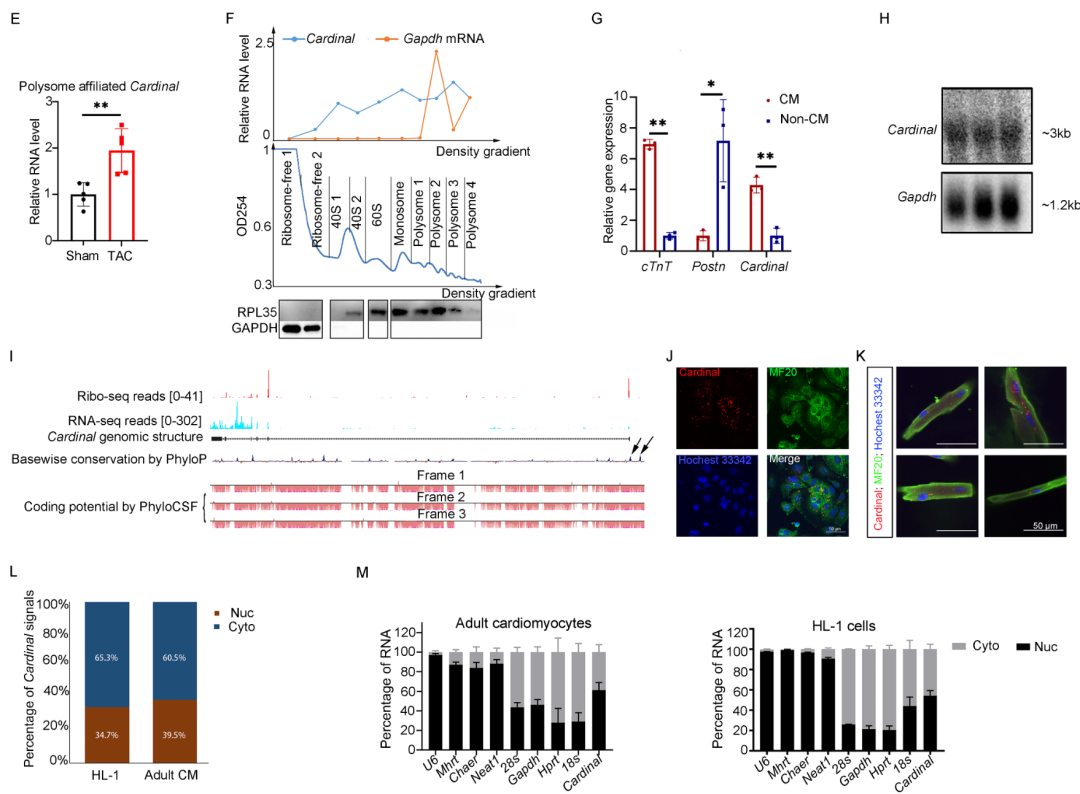
2. CARDINAL调控心肌细胞的蛋白翻译
下一步,探究CARDINAL对心肌细胞翻译的影响。首先通过嘌呤霉素摄取试验评估细胞的翻译速率。研究发现在大鼠乳鼠心肌细胞中,过表达CARDINAL并不影响生理状态下的翻译速率,但可以抑制苯肾上腺素(Phenylephrine, PE)刺激导致的翻译增强。另一可视化翻译量化方法FUNCAT也支持了该结果。进一步在成年小鼠心肌细胞中重复了该研究,结果同样提示CARDINAL过表达可以抑制PE导致的成年心肌细胞翻译增强。
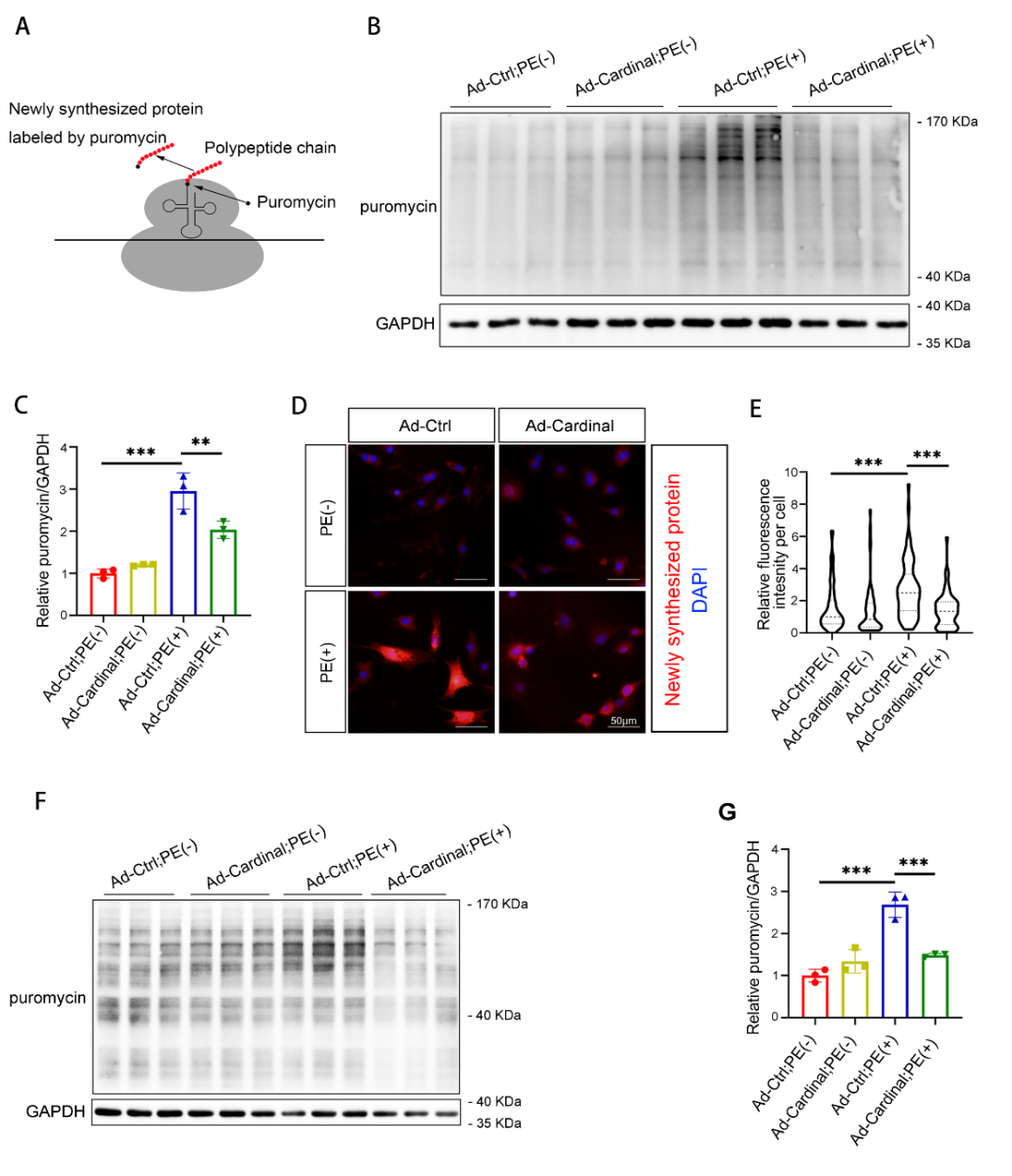
3. 核糖体结合的CARDINAL在心肌肥厚时上调
在初步明确CARDINAL对心肌翻译的调控作用后,进一步评估了CARDINAL与心肌肥厚的相关性。分析RNA测序数据库发现,CARDINAL在多种病因导致的心力衰竭中均上调,该结论在人和小鼠心力衰竭样本中的qPCR分析中得到证实。在成年小鼠心肌细胞中,PE刺激也可以导致CARDINAL升高。这些结果提示,CARDINAL升高可能参与了心肌肥厚的发生过程。CARDINAL的启动子区域在心肌肥厚时H3K9Ac修饰显著增强,提示其启动子在心肌肥厚时激活。对人和小鼠CARDINAL启动子区域的序列进行分析,提示MEF2和NFAT可结合CARDINAL启动子。通过MEF2A的CHIP-seq证实了MEF2与CARDINAL启动子结合。钙调磷酸酶A(Calcineurin A, CnA)是NFAT的上游激活因子,研究发现在CnA过表达小鼠的心脏中,CARDINAL上调约15倍,也支持了NFAT可以结合CARDINAL启动子这一猜想。最后研究分析了小鼠心肌肥厚样本的RNA-seq和Ribo-seq数据库,结果发现,心脏中的总CARDINAL在压力过负荷2天后即开始明显升高,而核糖体结合的CARDINAL在压力过负荷2周后才明显升高。这些结果提示,CARDINAL与核糖体的结合是受到动态调控的而非被动的非特异性过程。
4. CARDINAL基因敲除加重压力过负荷诱导的心肌肥厚
为了进一步探究CARDINAL对心肌肥厚的调控作用,构建了CARDINAL基因敲除小鼠,qPCR证实了CARDINAL被成功敲除。表型分析提示CARDINAL基因敲除并不影响生理条件下的心功能和心脏表型。但在主动脉缩窄术(Transverse aortic constriction)诱导的心肌肥厚模型中,CARINDAL敲除显著增加心脏重量、心脏大小、心肌细胞横截面积、心肌纤维化、上调心肌肥厚和心肌纤维化标志物和恶化心功能。通过在体嘌呤霉素摄取试验,证实了CARDINAL敲除可以促进心肌肥厚中的心脏翻译功能。以上结果表明CARDINAL基因敲除可以恶化心肌肥厚,该功能可能与翻译增强相关。
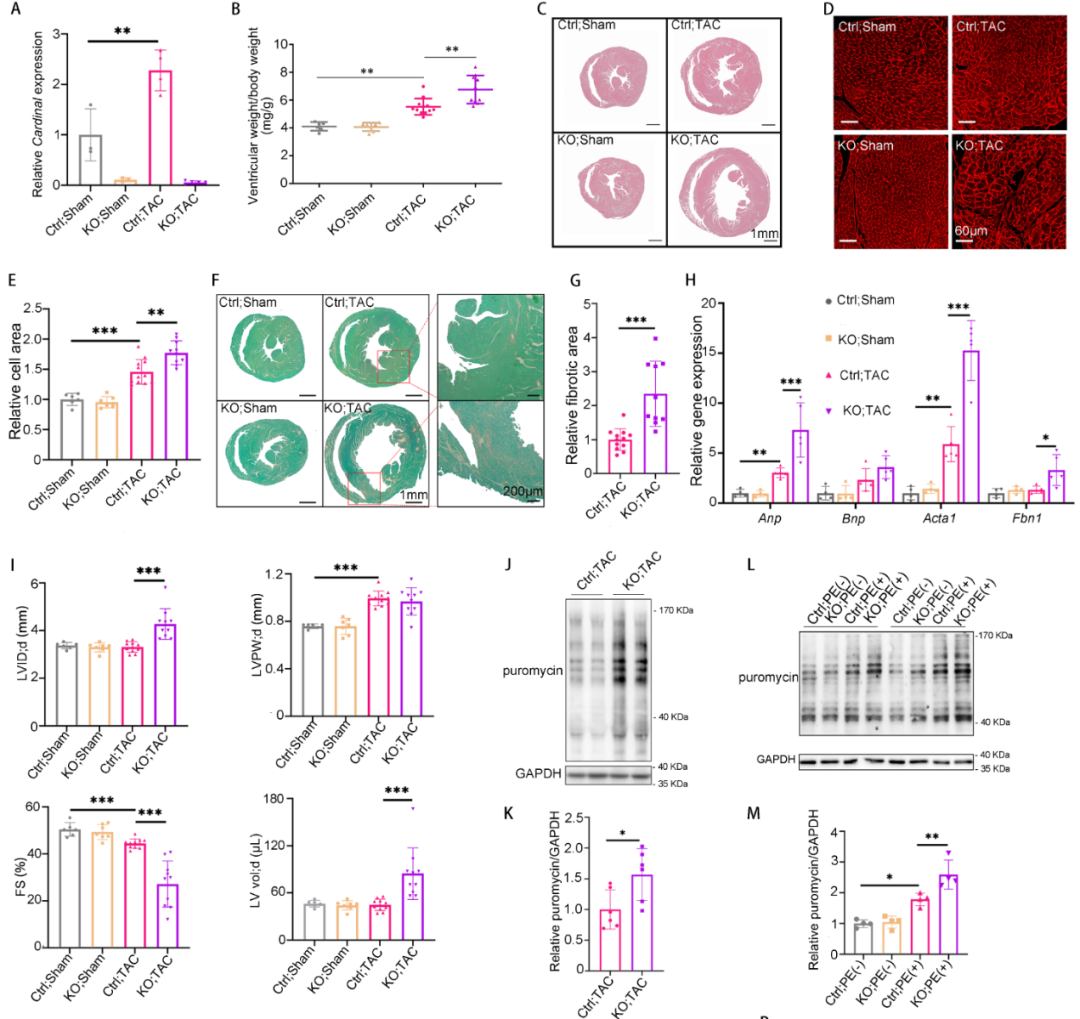
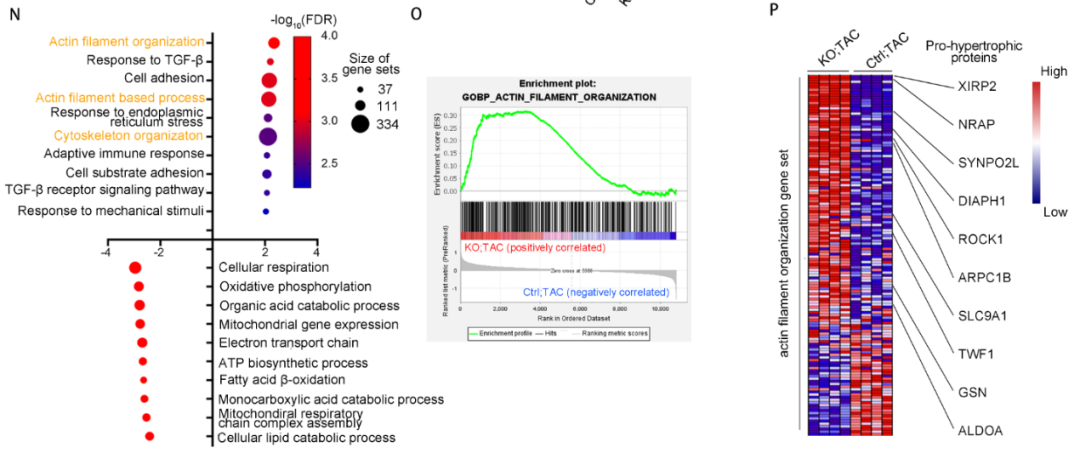
CARDINAL对蛋白的调控是否具有偏好性呢?研究通过定量质谱分析了CARDINAL-KO和对照小鼠TAC术后的蛋白组学。结果发现CARDINAL-KO可导致细胞骨架相关的蛋白显著上调,其中“肌动蛋白纤维组装”这一基因集上调最明显。研究通过Ribo-seq分析证实了蛋白组中“肌动蛋白纤维组装”基因集上调的确是由翻译增强导致的。进一步分析该基因集,研究发现了多个蛋白已被报道可促进心肌肥厚的发展。这一结果提示CARDINAL对调控细胞骨架蛋白翻译具有一定偏好,这一偏好性可解释其对心肌肥厚的调控作用。
5. CARDINAL过表达抑制压力过负荷诱导的心肌肥厚
下一步,研究拟探索CARDINAL过表达对心肌肥厚的治疗价值。通过构建重组AAV9病毒和小鼠皮下注射,成功在心脏中过表达了CARDINAL。同样,CARDINAL过表达不影响生理状态下的心脏表型。但在TAC诱导的心肌肥厚模型中,CARDINAL过表达可以减少心脏重量、心脏大小、心肌细胞横截面积、心肌纤维化、抑制心肌肥厚和纤维化标志物的上调、改善心功能。同样,在大鼠乳鼠心肌细胞构建的体外心肌细胞肥厚模型中,腺病毒介导的CARDINAL过表达可以抑制心肌细胞面积变大和肥厚标志物上调。以上结果提示CARDINAL过表达可以改善心肌肥厚。
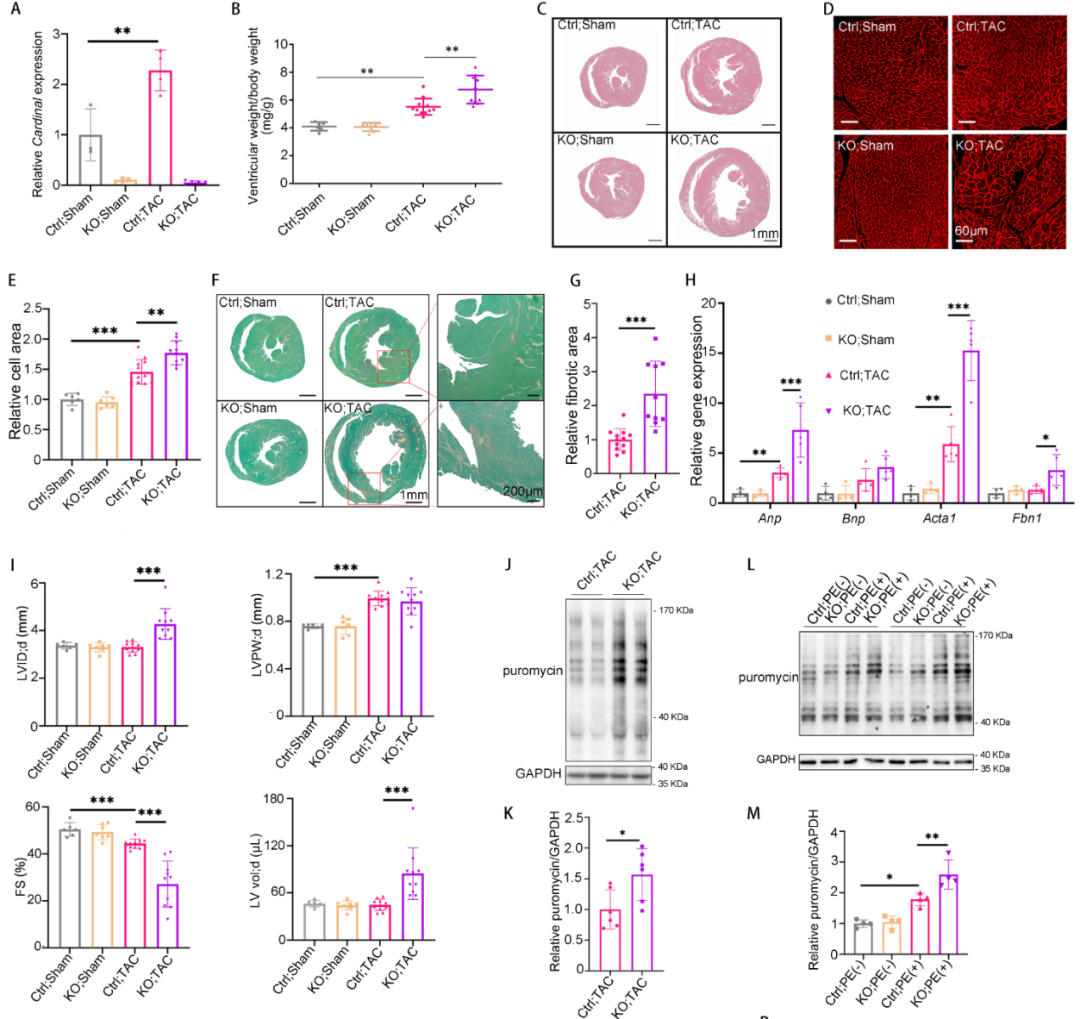
6.CARDINAL与翻译调控因子DRG1结合
为了探究CARDINAL调控翻译和心肌肥厚的具体分子机制,进行了3组CARDINAL下拉序贯质谱分析,3组质谱结果均提示CARDINAL可与Developmentally regulated GTP binding protein 1(DRG1)这个蛋白结合。体外的CARDINAL下拉序贯Western blotting和DRG1 RIP序贯qPCR实验均证实了两者可以互相结合。已有文献报道,DRG1结合于核糖体上,可以通过抑制核糖体停顿促进翻译。通过分析Ribo-seq数据发现,心肌肥厚过程中,MYH7基因的翻译存在明显的核糖体停顿过程,提示核糖体停顿很可能参与了心肌肥厚的发生和发展。
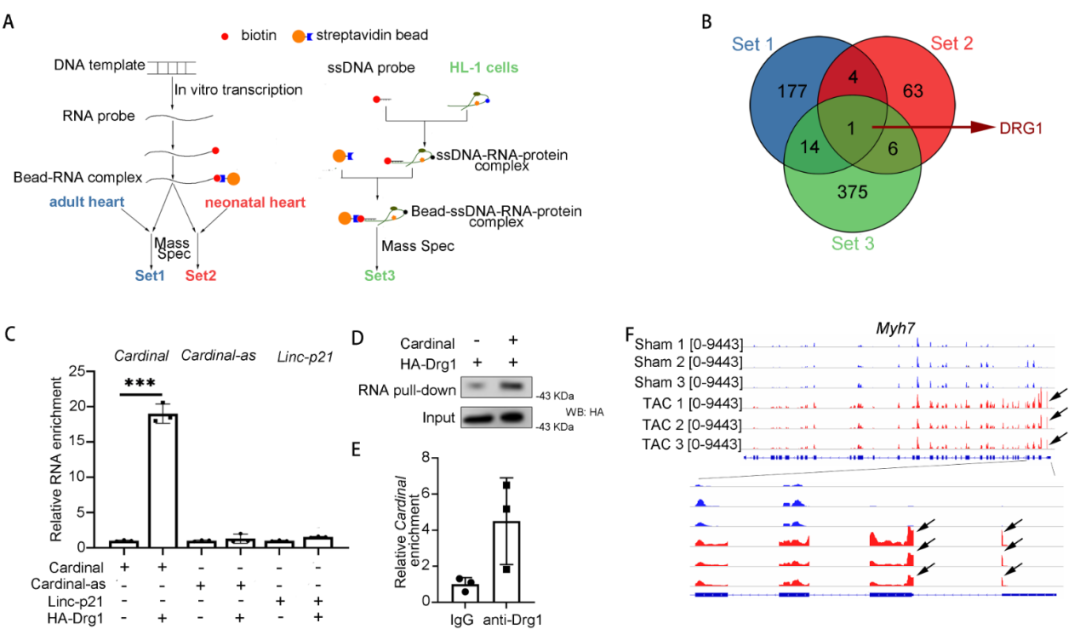
在HL-1心肌细胞系中发现DRG1敲降显著减慢翻译速率。而在大鼠乳鼠心肌细胞中,DRG1敲降显著抑制PE诱导的翻译增强,同时也抑制了PE诱导的细胞面积增大和肥厚标志物上调。以上结果提示DRG1可以通过促进翻译调控心肌细胞肥厚。
在HL-1心肌细胞系中发现DRG1敲降显著减慢翻译速率。而在大鼠乳鼠心肌细胞中,DRG1敲降显著抑制PE诱导的翻译增强,同时也抑制了PE诱导的细胞面积增大和肥厚标志物上调。以上结果提示DRG1可以通过促进翻译调控心肌细胞肥厚。
那么翻译停顿是否参与了CARDINAL对心肌肥厚的调控呢?首先通过查阅文献发现了4种氨基酸基序可以诱导核糖体停顿。于是再次分析了上述CARDINAL-KO和对照小鼠TAC术后心脏的蛋白组学结果,发现CARDINAL-KO小鼠心脏中显著上调的蛋白含有更多的核糖体停顿相关基序(无论是种类上还是数量上),这提示了CARDINAL-KO很可能是通过抑制核糖体停顿而促进翻译的。于是拟进一步探索,翻译停顿能否解释CARDINAL对细胞骨架相关蛋白调控的偏好性。于是研究再次分析了“肌动蛋白纤维组装”基因集中的蛋白,发现该基因集中的蛋白含有更多的核糖体停顿相关基序(无论是种类上还是数量上)。该结果提示细胞骨架相关蛋白由于含有更多停顿相关基序,所以其翻译更容易受到核糖体停顿的影响,所以在CARDINAL-KO导致的核糖体停顿抑制时,这些蛋白的翻译增强最为明显。
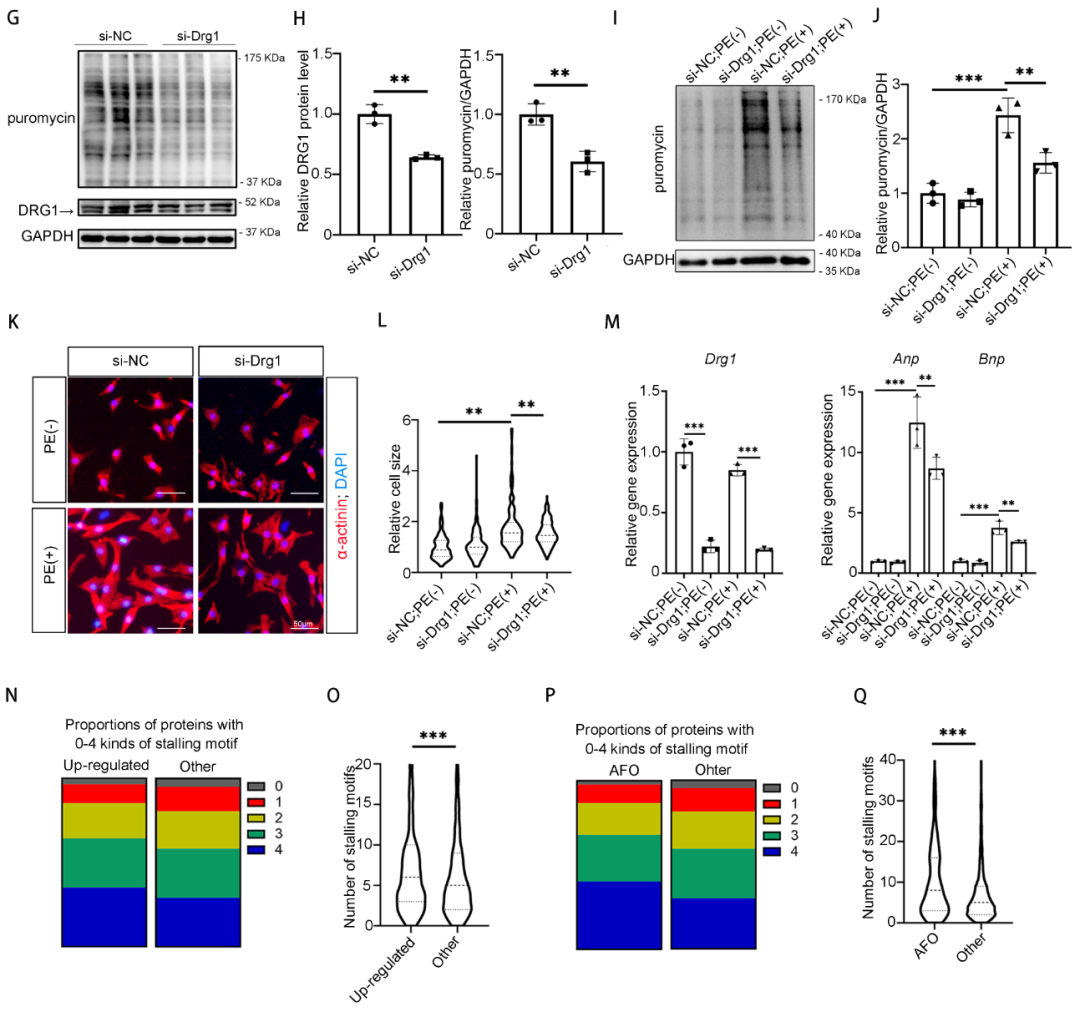
7. CARDINAL通过抑制DRG1-DFRP1结合降低DRG1的蛋白稳定性
下一步,拟探索CARDINAL是如何通过结合DRG1影响翻译的。无论在体内还是体外模型中,心肌肥厚中的DRG1蛋白表达量上调,而CARDINAL过表达可以抑制DRG1的上调,而CARDINAL敲除则会进一步促进DRG1蛋白的上调,但是DRG1的mRNA水平并没有类似的变化,这提示很可能是CARDINAL调控了DRG1的蛋白降解。已有研究表明,DFRP1是DRG1的结合蛋白,二者结合可以促进DRG1的蛋白稳定。研究通过免疫共沉淀(co-immunoprecipitation, co-IP)验证了二者的结合。进一步实验也验证了DFRP1过表达可以提高DRG1的蛋白含量。CARDINAL过表达可以抑制DFRP1过表达对DRG1蛋白含量的促进作用,提示CARDINAL可能可以抑制二者的结合。的确,在干预CARDINAL后进行co-IP实验发现,CARDINAL过表达可以抑制DRG1和DFRP1蛋白结合,而敲降CARDINAL则可促进二者的结合。最后,研究在PE诱导的心肌细胞肥厚模型中,敲降DRG1导致蛋白翻译抑制,但是在敲降DRG1的基础上进一步过表达CARDINAL则无法进一步抑制蛋白翻译,提示CARDINAL是通过下调DRG1蛋白含量调控心肌细胞翻译的。
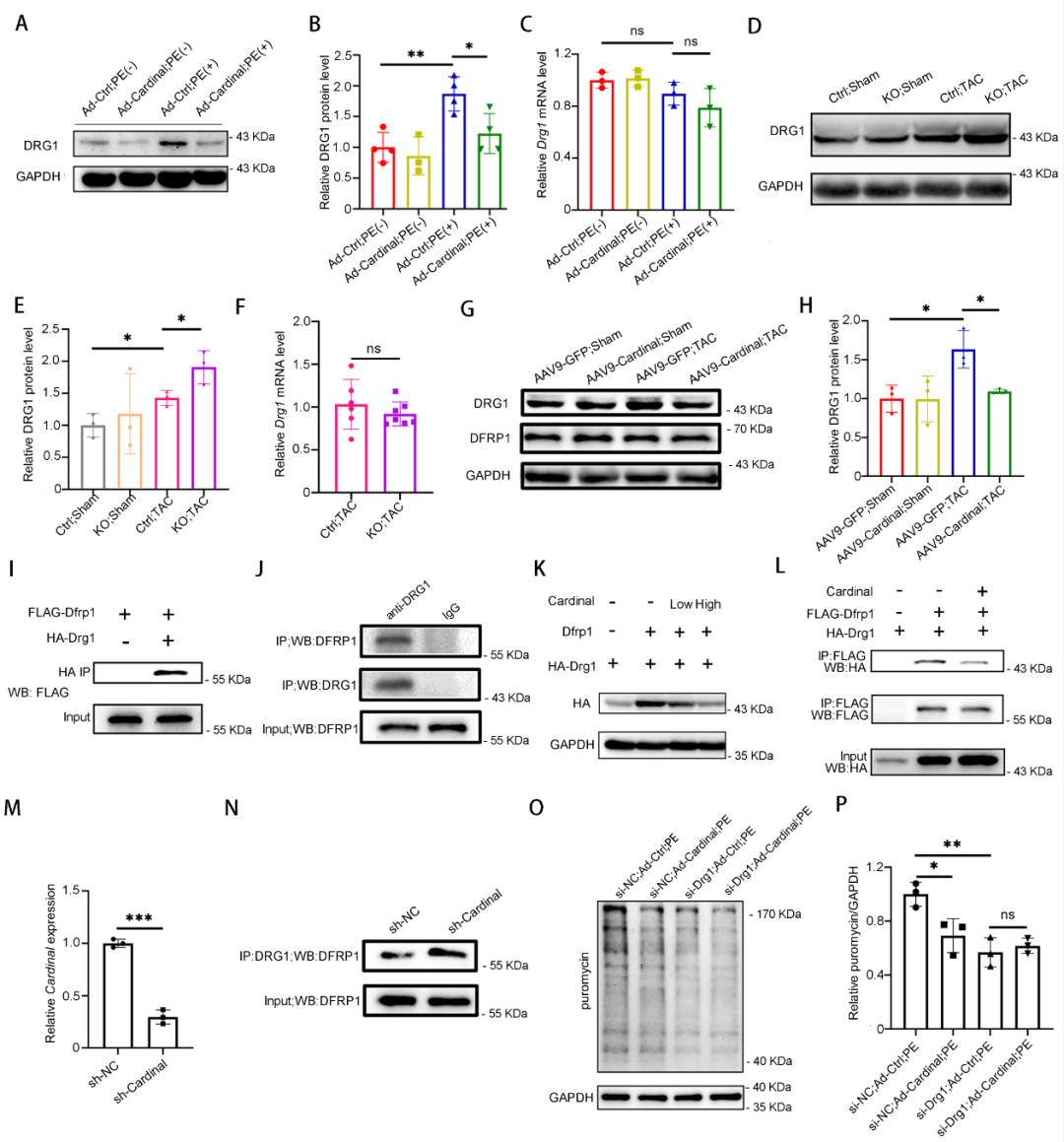
结论
本研究通过系统性的筛选发现了CARDINAL是心肌特异的核糖体结合蛋白,阐明了CARDINAL通过降低DRG1蛋白稳定性而抑制翻译,从而改善心肌肥厚的作用和其具体分子机制。该研究首次发现了核糖体结合lncRNA对核糖体翻译功能的调控作用,也为今后寻找心力衰竭的治疗靶点提供了全新的思路。
原文链接:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#心肌肥厚# #CARDINAL#
61