
没想到,多年的食欲不佳,病因竟是数十年前的祸根
2024-10-01 梅斯罕见新前沿 MedSci原创 发表于上海

8 岁伽罗食欲欠佳,经检查确诊海蓝组织细胞增生症。介绍其发病机制、临床表现、分类、辅助检查及诊断治疗,强调暂无有效疗法。
8岁的伽罗(化名)最近几年总是食欲欠佳,从当地村诊所一直看到省会三甲医院,一直没有找到原因,治疗效果不佳。这次慕名来到北京某三甲医院就诊,医生仔细看了孩子的病例:胃镜、B超都做了;血常规、生化、幽门螺旋菌也做了,除了脂代谢有点异常外,并未发现什么异常。
伽罗的症状很明确,却找不到病因。经过全院疑难病例会诊,医生建议:做骨髓检查。检验科医生观察标本时发现,一小堆的蓝色聚集在一起,幽幽地泛着光芒,像绿松石,所有视野下都有这种神秘蓝。海蓝组织细胞增生症确诊无疑。
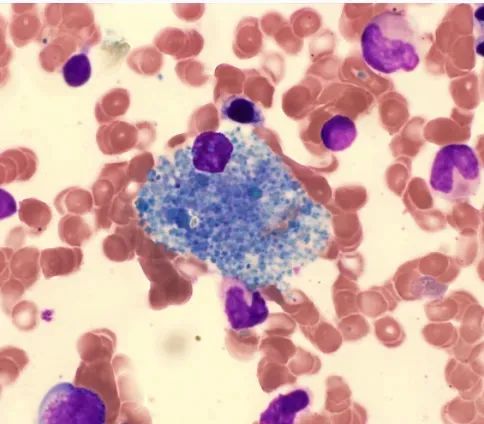
医生介绍,海蓝组织细胞增生症是一种遗传性代谢病,由于神经鞘磷脂酶活性降低,导致神经鞘磷脂和神经糖脂积累和异常代谢。病人先是脂肪代谢异常,然后消化系统功能紊乱。如果不进行治疗,可能会导致严重的健康问题,甚至危及生命。这种病发生率极低,全国从1980年到2006年文献记载仅确诊17例,目前并无有效的治疗办法。
伽罗为什么会得发病率这么低的罕见病?这病是常染色体隐性遗传性疾病,医生详细问了家族病史。原来女孩的爷爷和外婆有血缘关系,父母属于近亲缘结婚。
海蓝组织细胞增生症(SBH),又称海蓝组织细胞综合征,是一种少见的不典型的脂质贮积症。海蓝组织细胞可大量浸润骨髓、肝、脾、胃肠道、肺、脑及淋巴结等器官。1970年首先命名。因其组织细胞可以用瑞氏染色染成海蓝色而得名。
发病机制
有学者认为海蓝组织细胞是吞噬细胞本身结构(如线粒体)的异常所致的某种酶的缺陷,因而影响脂质的进一步分解,造成组织细胞中脂质的大量堆积,从而引起了一系列病理改变。亦有人认为,SBH可能是尼曼-匹克病(NPD)的一种变异型。
临床表现
起病年龄不限,从婴儿到老年都有可能发病。40岁以下发病的多见。该病征临床过程类似戈谢病慢性型,起病隐匿,病程长,均有肝脾肿大。脾大一般超过肝脏,浅表淋巴结多无肿大,因血小板减少,皮肤可见紫癜,少数病人有黄疸,可能因磷脂或糖脂在肝内蓄积引起进行性肝功能衰竭,偶可发生肝硬化。尚有皮肤出现色素,皮疹,眼底斑点区有白色环。1/3的病例有肺部浸润,像肺结核或结节病。
大部分患者的骨髓及肝、脾中可见海蓝组织细胞增生,1/3患者的肺组织中可见海蓝组织细胞。继发于CML、PV、ITP、慢性肉芽肿性疾病、结节病、SLE、淋巴瘤、珠蛋白生成障碍性贫血、镰状细胞贫血、高脂蛋白血症、戈谢病、尼曼-匹克病、多发性骨髓瘤、Wolman′s病及肝硬化等疾病者称为继发性SBH,继发性SBH可能与酶系统异常或脂质代谢负担过重有关。
四个形态
海蓝组织细胞分为4种形态:
Ⅰ型(颗粒型):胞质内可见大而均一的深蓝色颗粒,其直径约3~ 4 μm,多少可不等,可充满胞浆,泡沫不明显。
Ⅱ型(泡沫型):胞质内呈泡沫状,胞壁较厚,无色或个别区域染成粉红色,内含红色至浅紫色或深紫色物质。
Ⅲ型(颗粒泡沫型)介于Ⅰ~ Ⅱ型之间的中间型,在泡沫状胞质中有数量不等的蓝色或深蓝色颗粒。
Ⅳ型(退化型)系Ⅱ ~ Ⅲ型SBH衰老退化形成,该型胞质不清楚,胞核结构模糊。
三个分类
根据起病原因,海蓝组织细胞增多症可以分为三类:
(1)原发性。完全排除继发因素,无明显遗传倾向的先天性神经鞘磷脂酶缺陷
(2)原发性家族性。家系调查符合常染色体隐性遗传性疾病的SBH,目前研究提示可能与apo基因突变相关;
(3)继发性。继发于某些疾病,可能与酶系统异常或者脂质代谢负担过重有关,目前比较明确的继发疾病主要有CML、PV、ITP、MDS、恶性淋巴瘤、肝硬化、多发性骨髓瘤、结缔组织病、遗传性溶酶体病、地中海贫血、脾功能亢进、某些感染疾病等。
以上各种原因会导致组织细胞内蜡样质、神经鞘磷脂和神经鞘糖脂积聚,使得组织器官肿大,最终导致其功能降低或丧失。
辅助检查
1、血象:血红蛋白、白细胞计数正常,血小板减少。
2、骨髓、红、粒、巨核系增生正常,可见海蓝组织细胞(I型)和泡沫细胞(II型)。海蓝细胞直径20~60μm,有一偏位圆形核,染色质凝集,可见核仁,胞浆含不等数量的海蓝色或蓝绿色颗粒,其苏丹黑及糖原染色呈阳性反应,浆内含有脑苷脂和糖类物质。电镜下显示类脂分子呈圆周状板层结构。
3、病理检查 对肝与脾的提出物分析,结果糖神经鞘酯、磷脂和脑苷脂增加,部分病人在肝、脾、肺组织中也有海蓝细胞浸润。
4、b超 可见肝、脾肿大,肝硬化表现。
5、x线胸片 见肺部浸润影。
6、脑电图检查 可发现异常脑波。
诊断与治疗
目前该病的确诊依赖于骨髓、肝、脾、肺等组织检查发现海蓝组织细胞,在确定海蓝组织细胞综合征后,尚须进一步寻找原因,在逐一除外继发性之后,才能确诊为原发性海蓝组织细胞综合征。
骨髓活检标本瑞吉染色显示镜下海蓝组织细胞,胞体较大,外形不规则,细胞核偏位,胞质较丰富,呈蓝色粗颗粒状、部分大小不一。
糖原染色呈阳性,可与组织细胞吞噬含铁血黄素鉴别。
镜下发现海蓝组织细胞是确诊海蓝组织细胞增生症的重要确诊条件之一。
本病暂无有效的治疗方法,有脾大及脾功能亢进者可行脾切除术,但仅能缓解症状,不能解除病因,且手术治疗还有可能加速高脂血症和肝脏脂质沉积,需严格掌握本病手术指征。
参考文献:
[1]孙涛,张丹丹,赵怡雯,海蓝组织细胞增生症1例并文献复习[J]。武警后勤学院学报,2015,24(12):996-7。
[2] 杨玉姣,茅彬等。13个白化病家系的遗传学分析及产前基因诊断[J]。中华遗传学杂志,2022,10(2),143-7。
[3]金晓希.224例慢性髓系白血病伴类脂质细胞增多患者骨髓检测结果分析[J].国际检验医学杂志,2012,33(22):2768-2769
[4]谢易,傅君芬,梁黎.原发性海蓝组织细胞增多症1例[J].中国实用儿科杂志,2013,(1):74-75.
[5]赵倩倩,刘力,马继军,胡坚.海蓝组织细胞增生症1例[J].中国中西医结合外科杂志,2015,(1):82-83.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#罕见病# #海蓝组织细胞增生症#
47