
散发性甲状腺髓样癌的年轻患者基因检测有哪些发现?常见捣蛋基因少不了它
2024-04-29 苏州绘真医学 苏州绘真医学 发表于陕西省

本研究表明,在早发性非遗传性MTC队列中,RET仍然是主要的驱动基因。在RET阴性肿瘤中,NF1和RAS是散发性MTC的驱动因素。
对散发性甲状腺髓样癌(MTC)进行的基因分析显示了RET、RAS以及偶尔其他基因的体细胞变异。然而,约20%的散发性MTC患者缺乏已知的基因驱动因子。为了发现潜在新的体细胞或胚系驱动因素,研究者分析了一组独特的散发性、极早发性和侵袭性MTC患者。19例患者进行了胚系和体细胞DNA外显子组测序,此前这些患者的胚系RET变异检测呈阴性。
对19份胚系样本进行的外显子组测序证实了不存在RET变异,并在1例患者中发现了NF1致病性变异。15例肿瘤的体细胞测序成功,结果显示80%的肿瘤有RET变异,主要是与疾病侵袭性相关的p.Met918Thr变异。在RET阴性的肿瘤中,发现了HRAS和NF1的致病性变异。研究者在既往未被临床诊断为1型神经纤维瘤病的患者中观察到NF1胚系和体细胞变异,这表明NF1的杂合性缺失可作为潜在的MTC驱动因素。体细胞拷贝数变异(SCNA)分析显示,53.3%的肿瘤有染色体变异,主要是在RET阳性病例中,其中9号染色体和22号染色体缺失最常见。
本研究表明,在早发性非遗传性MTC队列中,RET仍然是主要的驱动基因。在RET阴性肿瘤中,NF1和RAS是散发性MTC的驱动因素。此外,对于无RET胚系突变的年轻患者,在考虑胚系NF1基因分析的基础上进行仔细的临床评估是排除1型神经纤维瘤病(NF1)的理想选择。
研究背景
甲状腺髓样癌(MTC)有2种原发形式:散发性(75-80%)和遗传性(20-25%)。遗传性MTC与2型多发性内分泌腺瘤(MEN2)相关,这是一种常染色体显性综合征,根据临床特征分为MEN2A或MEN2B。MEN2A涉及MTC、嗜铬细胞瘤和甲状旁腺功能亢进。相反,MEN2B(较罕见的形式,5%)具有明显的早发、侵袭性MTC、嗜铬细胞瘤、黏膜神经瘤和马方综合征样表型。遗传性MTC的标志是存在胚系RET原癌基因致病性变异。
散发性MTC通常在40-60岁发病,体细胞RET变异是主要的癌基因(在未经选择的队列中占40%-50%,在晚期病例中高达85%)。在RET变异中,p.Met918Thr最为常见,且与侵袭性病程和不良预后相关。较少见的RET变异包括密码子611、618、620、630、632、634、768、791和883的错义变异,以及小的插入缺失(indels)。HRAS和KRAS变异占散发MTC病例的20%-30%,最近报道了3例散发性MTC没有RET和RAS变异,而是以NF1的双等位基因失活为标志,导致已知激活RAS信号通路的肿瘤抑制基因的杂合性缺失(LOH)。COSMIC(癌症体细胞突变目录)数据库显示,相当大比例(约30%)的散发性MTC病例缺乏未知的驱动基因,目前的研究已经将其缩小到20%以下。
本研究探讨了一个无致病性RET胚系变异的早发性和侵袭性MTC患者的特定队列的分子谱。这一患者亚组引起了研究者的注意,原因是发病年龄早、缺乏胚系RET变异以及局部和远处转移性疾病的发生率高(分别为94%和68.4%),这与MEN2B患者队列相似(分别为100%和72.2%;P= .48和P= .85)。两组患者的疾病结构进展和系统性治疗需求也相似。
研究方法
研究者从270例MTC患者(包括101例散发性病例)中,选择了19例晚期、早发性和散发性MTC患者。考虑到散发性MTC通常在40-60岁之间出现,研究者将疾病早发性定义为在30岁之前被诊断出。研究者对原发肿瘤或当无法获得原发肿瘤时对转移性肿瘤进行了全外显子组测序,并从血液样本中提取匹配的DNA。
研究结果
患者的临床特征:
19例患者的中位年龄为21岁(范围:8-29岁)。19例患者中4例(21%)≤12岁,另外有4例(21%)在13-18岁。女性14例(73.7%)。所有患者在诊断时均有颈部淋巴结转移,其中10.5%(2/19)有远处转移。原发肿瘤中位大小为4 cm(范围:0.6-10.3 cm),26.3%(5/19)为多灶性,10.5%(2/19)存在C细胞增生区。中位随访时间为9.8年(范围:0.5-29年),在分析时,73.7%(14/19)有远处转移,其中47%(9/19)有≥3个转移部位。骨转移最常见(78.5%),其次为肺和肝转移(64.3%)。随访期间,10例(52.6%)患者出现影像学疾病进展,需要酪氨酸激酶抑制剂(TKI)治疗。2例患者分别于17岁和30岁因疾病进展死亡。研究者总结了临床和随访特征(表1和2)。
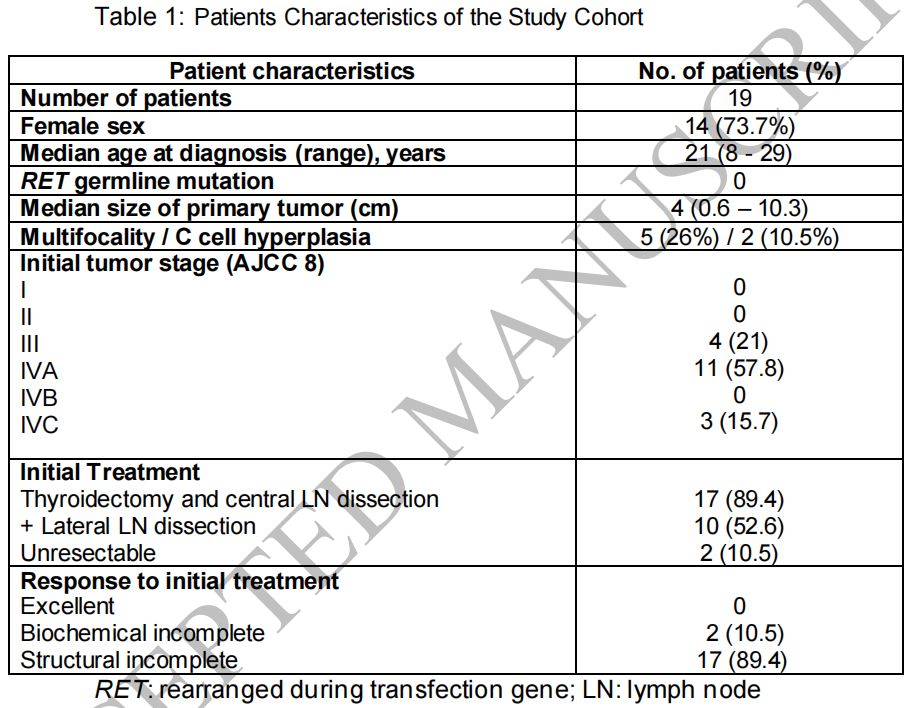
表1

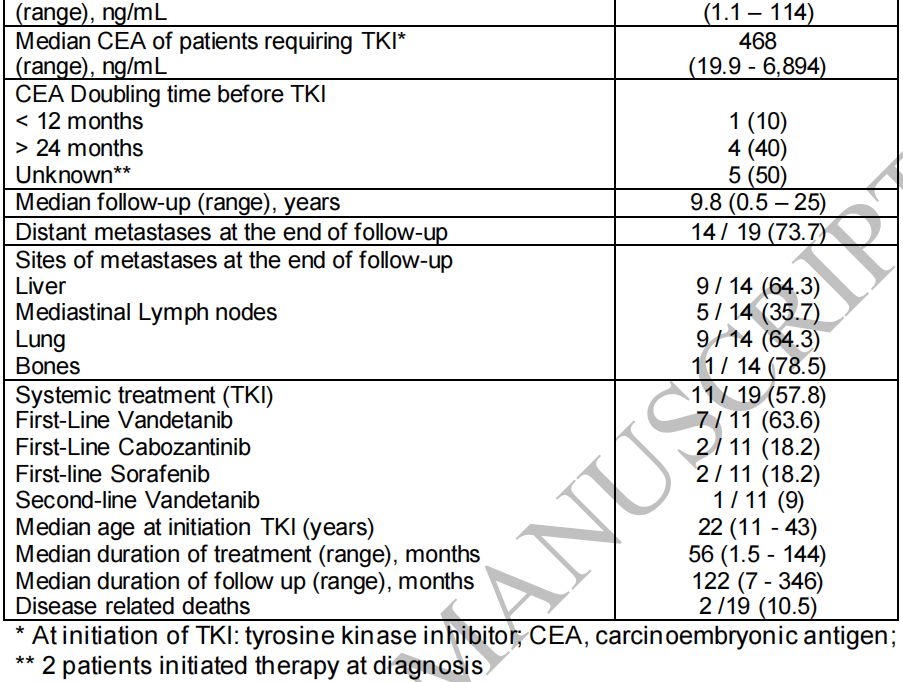
表2
胚系分析谱:
对19份胚系标本进行的外显子组测序显示,1例患者携带NF1变异(c.1527+1G>T),该变异被美国医学遗传学与基因组学学会归类为致病性,未在人群数据库(ExAC)中发现,并且与NF1相关(图1)。实验研究证实了其对mRNA剪接和蛋白功能破坏的影响。

图1
在该队列中未发现胚系RET变异,并且除NF1变异外,未发现其他致病性变异。在5例携带体细胞驱动基因的患者中观察到FOXE1、KAT6A、LZTR1、NTHL1、POLR2E和TERT等与肿瘤发生相关基因的意义不明变异(VUS)。另外,在2例肿瘤DNA无法获得的患者中发现了VUS(CDON和NTHL1)。
体细胞分析谱——单一变异:
研究者对18例获得的肿瘤进行了外显子组体细胞测序:3例因DNA质量差而被排除,只剩下15例肿瘤。40%(6/15)的甲状腺原发灶和60%(9/15)的淋巴结转移灶可提取DNA。大多数标本(93.3%,14/15)为福尔马林固定石蜡包埋,1例为新鲜冰冻淋巴结活检标本。
在本队列中,93.3%(14/15)的患者存在单个相互排斥的体细胞变异,其中1例未发现变异。RET是最常见的突变基因,占80%(12/15)。RET基因第16号外显子p.Met918Thr变异是最常见的体细胞变异,占60%(9/15)。在20%(3/15)的患者中观察到其他RET变异,包括COSMIC数据库中报告的2个(p.Cys630Arg和p.Arg873Trp),以及1个新的RET框内插入p.Glu632_Ser645dup(表3)。
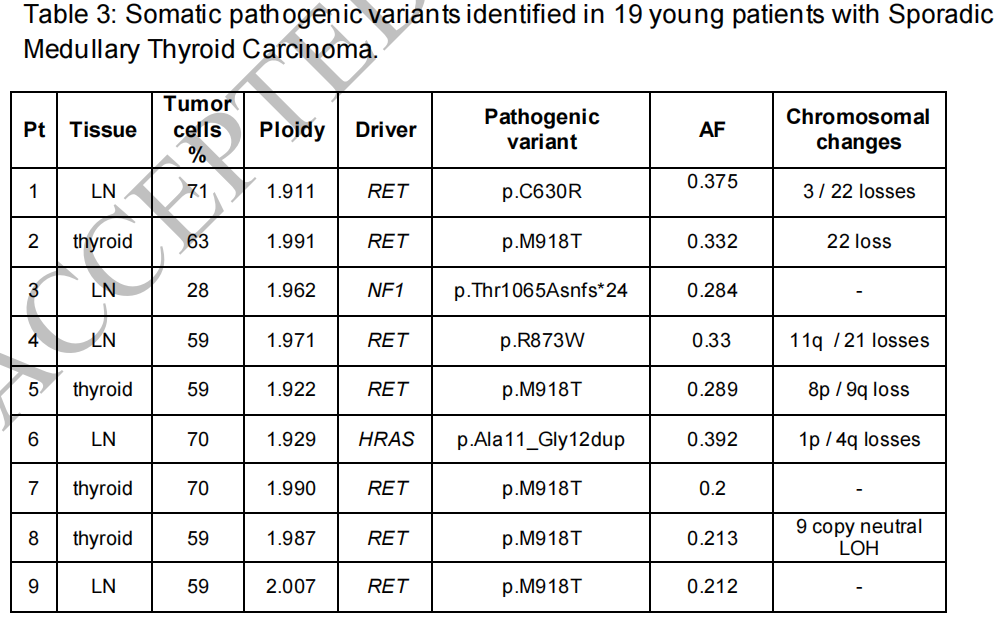

表3
仅检出1个HRAS致病性变异(6.6%),这是一个已知与MTC有致病性关联的框内插入变异(p.Ala11_Gly12dup)(表3)。该患者8岁时被诊断为MTC,经过15.6年的长期随访,生化疾病稳定[降钙素12.5 pg/mL,癌胚抗原(CEA)1.93 ng/mL]。
在携带胚系NF1变异(c.1527+1G>T)的同一患者中发现了一种新的体细胞NF1框内插入变异(p.Thr1065Asnfs*24),提示该肿瘤抑制基因的双等位基因失活(表3)。有趣的是,这名22岁女性因存在马方样体质但无其他MEN2B特征而被转诊为MEN2B疑似患者。评估未发现RET胚系变异,但发现3个小牛奶咖啡斑点提示存在未定义的遗传综合征。在就诊数年后发现胚系NF1变异后,进行了更详细的检查,发现12个牛奶咖啡斑点和腋窝雀斑,但没有神经纤维瘤。确诊时,术前血清降钙素3182 pg/mL。甲状腺全切除术和双侧中央区淋巴结清扫显示2 cm单发MTC伴C细胞增生和中央区淋巴结转移(T1bN1a)。患者一直处于生化不完全疾病状态,直到3年后发现颈部复发并进行了颈淋巴结清扫术。诊断后7年,患者检出肝转移,但疾病保持稳定,并在监测中。
体细胞分析谱——SCNA 分析:
通过SCNA分析(包括全染色体或单臂改变)评估的肿瘤染色体不稳定性影响了53.3%(8/15)的病例,详细信息见表3。
基于体细胞RET变异分析SCNA检出率,在12例RET阳性肿瘤中,7例(58.3%)为SCNA,而在3例RET阴性肿瘤中,1例为SCNA。携带NF1变异的肿瘤未显示SCNA。在RAS阳性肿瘤中,2例部分缺失发生在1号染色体和4号染色体。在7例RET阳性的SCNA肿瘤中,有17个染色体缺失,1个染色体增加,1个拷贝中性LOH事件。受累染色体数各不相同:2例为1条,4例为2条,1例为多个单体(染色体1p、3、4、5q、9和13)。最常见的缺失发生在9号和22号染色体,在SCNA阳性肿瘤中均占比37.5%(3/8),仅发生于RET阳性病例。
体细胞检查结果与预后的相关性:
研究者将体细胞RET和(或)SCNA的存在与几个预后特征进行了关联:诊断时的TNM、随访期间发生远处转移、需要全身性治疗或死亡。在这个由年轻患者和侵袭性疾病组成的队列中,仍然可以发现RET变异的存在与需要全身性治疗之间的显著相关性(P= .03;V=0.53)。然而,无论是否存在RET变异,研究者发现每个肿瘤的SCNA数量或存在22号和9号染色体缺失与较差结局之间不存在显著相关性(P=0.77)。
尽管在本队列中没有观察到SCNA与不良预后的显著相关性,但1例表现为多个染色体缺失(1p/3/4/5q/9和13)引起了研究者的关注。一名RET 918 MTC的16岁患者(患者19,表3)出现局部晚期疾病和疼痛的广泛骨受累,且CEA与降钙素水平不成比例地升高,分别为79 pg/mL和4999 ng/mL,提示疾病去分化。尽管接受了骨放疗、冷冻消融和凡德他尼治疗,患者仍继续进展,并在6个月内死亡(图2)。
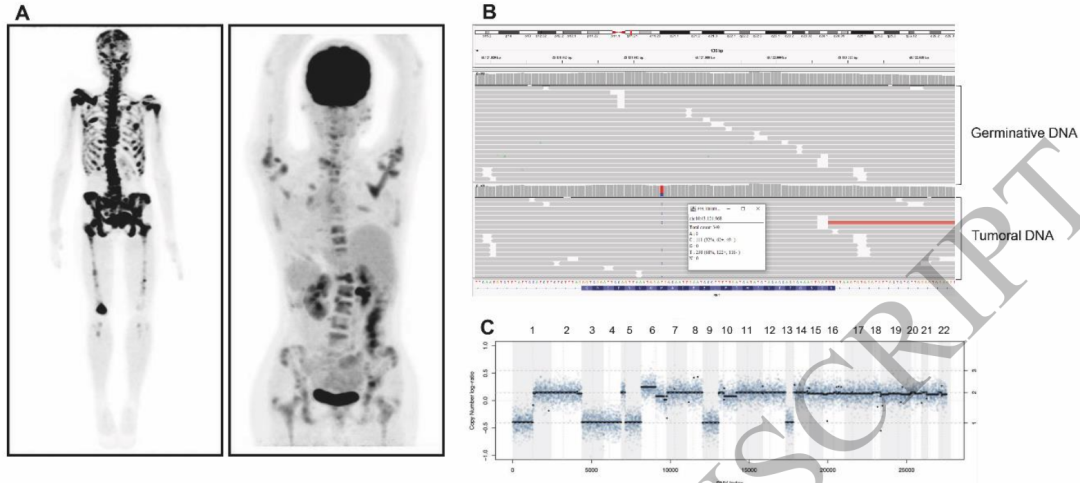
图2
携带22号染色体SCNA患者的临床和病理特征:
3例患者携带RET单个体细胞变异(p.Cys630Arg、p.Met918Thr和p.Glu632_Ser645dup),均表现为22号染色体完全缺失。其中2例分别在18岁和29岁时诊断为MTC,表现为颈部侧方淋巴结转移。4年和15年后,由于累及3个以上部位的进展性转移,患者开始接受索拉非尼治疗(一种RET、RAF激酶和血管内皮生长因子抑制剂)。值得注意的是,他们表现出了持久的应答,治疗时间超过了 10 年。第三例患者在15岁时被诊断为MTC(T2N1a),3年后发展为稳定的转移性肝病,并且迄今保持稳定。
讨 论
为了更深入地了解散发性MTC的发病机制,研究者建立了一个主要由年轻的非遗传性MTC患者组成的丰富队列。这些个体是从270例MTC患者(其中101例为散发疾病)的更大队列中选择的。在散发病例中,19例患者在30岁之前被诊断,其中8例在18岁之前被诊断。所有患者表现出与MEN2B相似的侵袭性临床特征。这个队列是一个独特的群体,研究者认为更有可能识别出不同的基因变异。
在本研究中,研究者分别对19例和15例患者进行了胚系和体细胞外显子组分析。胚系检测证实不存在胚系RET变异,并显示一个NF1致病性变异(c.1527+1G>T)。未发现其他胚系致病变异。
在15例具有肿瘤谱的患者中,93.3%(14/15)存在单一且相互排斥的体细胞变异。在1例同时分析了血液和肿瘤样本的病例中,广泛的分析未能发现明确的驱动因素。在已知的引起MTC的体细胞致病变异中,HRAS的发生率为6.6%(1/15),而RET变异的发生率为80%(12/15)。值得注意的是,在1例无体细胞RET和RAS的患者中,发现了胚系和体细胞NF1致病变异。
在12例携带RET基因体细胞变异的患者中,p.Met918Thr最常见,占75%(9/12),25%(3/12)的患者携带其他RET基因体细胞变异(p.Cys630Arg、p.Arg873Trp和1个新的框内插入p.Glu632_Ser645dup)。在文献中,散发性MTC的体细胞RET变异是参与肿瘤发生的最常见的驱动癌基因,在晚期病例中高达85%。显然,最常见的RET体细胞变异是p.Met918Thr,在包括原发肿瘤大小超过2 cm和晚期疾病患者的研究中,该变异的发生率较高。研究者在本队列中验证了这一发现,确定RET是主要驱动癌基因,即使在患侵袭性疾病的年轻患者中也是如此,正如研究者在80%的病例中观察到的那样。值得注意的是,p.Met918Thr致病性变异最常见,占本研究RET病例的75%。在这个队列中,研究者能够证明任何体细胞RET变异的存在与需要全身性治疗的疾病进展显著相关,正如之前在非选择的MTC队列中观察到的那样。
关于SCNAs,研究者未发现每个肿瘤的SCNA数量或22号染色体和9号染色体缺失与较差结局之间有显著相关性,且与RET变异的存在无关(P=0.77)。在本研究中,53.3%(8/15)的肿瘤存在SCNA。如之前报道的,最常见的是染色体9和22缺失,仅发生于RET阳性肿瘤。之前已经观察到3号、9号、10号和16号染色体的改变与不良预后相关,但未观察到22号染色体的SCNA改变与不良预后相关。
本研究未发现9号染色体改变与不良预后之间的相关性,这可能是因为本队列主要由侵袭性疾病患者组成。然而,在本研究这例侵袭性最强的患者中,肿瘤分析显示除了多个其他染色体(1p、3、4、5q和13)缺失之外,9号染色体完全缺失。患者16岁时出现低分化MTC,局部和全身治疗失败,迅速进展至死亡(图2和表3)。
在Ramone等人的研究中,22号染色体缺失与不良预后无关,这与本研究的发现一致。22号染色体包含多个肿瘤抑制基因,如LZTR1、NF2、CHEK2和SMARCB1。事实上,导致LZTR1失活的致病变异与Ras/MAPK通路的强烈激活有关,从而导致诸如神经鞘瘤病和RASopathies等疾病。有趣的是,在本队列中,3例22号染色体缺失的患者中有2例对索拉非尼有持续应答,索拉非尼是一种多激酶抑制剂,除作用于RET、VEGFR、PDGF和c-Kit外,还对Raf-1和B-Raf有强效抑制作用。这些患者对索拉非尼的应答维持时间超过10年。根据研究者的经验,这一结果值得注意,侵袭性转移性疾病患者对索拉非尼的应答较差。因此,需要提出的问题是,22号染色体缺失是否可以作为Raf抑制剂治疗应答良好的标志物。
在携带胚系NF1致病性变异的同一患者中发现了一个新的框内插入变异NF1(p.Thr1065Asnfs*24),强烈提示该肿瘤抑制基因的双等位基因失活。1型神经纤维瘤病(NF1)是一种多系统疾病,其特征为皮肤咖啡牛奶斑点、间质雀斑、多发性皮肤神经纤维瘤、皮下或深部结节性神经纤维瘤、丛状神经纤维瘤和特征性眼部体征。NF1许多临床特征的频率随着年龄的增长而增加,并且一些成年后明确患有NF1的患者无法在儿童早期(NF1特征变得明显之前)得到诊断。MTC与NF1相关,但到目前为止,尚不清楚这些肿瘤是否由NF1基因的二次打击引起。很少有报告表明MTC与NF1相关,虽然大多数报告未包括胚系或体细胞基因分析,但至少有2项研究在这类患者中发现了RET和NF1胚系变异。
最近,Shi等人在1例诊断为NF1的患者的MTC肿瘤中发现了致病性体细胞NF1变异。在此报告之后,研究者对2例非遗传性、非RET、非RAS MTC患者的NF1基因进行了分析,发现了NF1体细胞致病变异:1例确诊为NF1的患者有1个变异,另1例非NF1患者有2个获得性体细胞NF1基因变异。这些发现支持NF1在MTC肿瘤发生中的致病作用。神经纤维蛋白(一种已知可下调Ras通路的蛋白)功能的丧失可激活Ras/MAPK和PI3K/AKT/mTOR通路的GTPase活性,这可能解释了其在MTC中的驱动作用。
既往对散发性MTC进行的全外显子组研究因缺乏关于肿瘤致病变异与临床和病理特征之间相关性的数据而受到限制。本研究为文献提供了有价值的见解,揭示了具有侵袭性和早发性疾病特征的散发MTC患者队列的分子谱。这些分子发现以及患者的临床数据可能对个体化治疗和优化患者管理具有潜在的临床意义。
在目前有效的RET特异性抑制剂的情况下,RET突变频率高的证明,以及Ras-MAPK通路在肿瘤发生和生长中的重要性(在所分析的肿瘤中,观察到93.3%在该通路中有驱动变异),巩固了通过肿瘤体细胞分析确定晚期年轻患者最佳治疗模式的重要性。此外,本研究的发现提示了SCNAs作为治疗反应标志物的潜在作用。
研究者承认本研究存在局限性,特别是尽管纳入了一个特别独特、罕见和不常见的队列,但样本量相对较小。研究者同时使用了原发肿瘤和淋巴结样本(尤其是在没有原发肿瘤样本的情况下),并且大量使用了福尔马林固定的石蜡包埋组织样本,这可以归因于本研究是回顾性的。此外,认识到外显子组测序分析的局限性非常重要,尤其是与基因组测序相比,外显子组测序分析在分析重排方面的能力,以及在检查调控区和深层内含子区域方面的局限性。
尽管有这些局限性,但研究者相信本研究做出了重大贡献,尤其是因为它是第一个专门针对侵袭性疾病年轻患者的研究。鉴于研究者决定专门选择一个类似于MEN2B患者的队列(包括患有侵袭性疾病的年轻人),因此在分析分子异常和疾病侵袭性行为之间的相关性时,样本量固有地有限。
本研究证实,即使在非遗传性的年轻MTC患者中,RET也是主要的基因驱动因素(80%),并且表明在RET阴性肿瘤中,除了RAS之外,NF1也是一个新的驱动因素。在既往无NF1诊断或家族史的年轻患者中发现胚系和体细胞NF1变异时,应警惕进行全面评估以排除NF1。在本研究中,SCNA很普遍,并且主要发现在RET阳性肿瘤中。在这一有限的侵袭性疾病患者队列中,研究者未能证明与不良结局的相关性,但发现了22号染色体缺失作为TKI治疗反应标志物的潜在作用。这些发现的临床意义尚不清楚,但值得进一步研究。
参考文献:
Luciana Audi Castroneves, Flavia Regina Rotea Mangone, Antonio Marcondes Lerario, Ana Maria da Cunha Mercante, Rafael Loch Batista, Luciana Rodrigues Carvalho Barros, Carla Vaz Ferreira, Evelin Cavalcante Farias, Felipe Augusto Brasileiro Vanderlei, Ana Luiza Maia, Maria Aparecida Nagai, Alexander Augusto Lima Jorge, Ana Oliveira Hoff, Not Only RET but NF1 and Chromosomal Instability Are Seen in Young Patients with Sporadic Medullary Thyroid Carcinoma, Journal of the Endocrine Society, Volume 8, Issue 6, June 2024, bvae059, https://doi.org/10.1210/jendso/bvae059

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#甲状腺髓样癌# #散发性甲状腺髓样癌# #MTC#
0