
遗传的蛋白尿?一家4口患有同一种罕见病,等待他们的是透析和肾移植
2024-09-13 梅斯罕见新前沿 梅斯罕见新前沿 发表于上海

15 岁超超视力听力下降且有蛋白尿,被诊断为 Alport 综合征,介绍其症状、分类、诊断及治疗,其家人也患病。
15岁的超超(化名)最近5年视力下降的厉害,最近两年听力也有所下降,最近体检时还发现有蛋白尿。虽然这些年辗转多地求医,治疗效果却一直不太好。这次超超父母慕名带着他来到某医院就诊。
接诊医生先为超超做了眼部检查,裸眼视力右眼0.1,左眼0.3;矫正视力右眼0.7, 左眼0.7;双眼晶状体前表面中央区局限性前突, 呈锥形,囊膜下可见小斑点状白色混浊;双眼后极部黄斑区周围可见散在的黄白色点状颗粒;眼底血管荧光造影检查示双眼黄斑拱环结构破坏。随后的耳科检查也不容乐观,耳科电测听:双耳对称性感音神经性耳聋( 中、重度)。同时尿蛋白53mg/ L,血尿素氮7.3 mmol/ L,血清肌酐187.6 μmol/ L。
医生怀疑超超患有一种遗传性肾病,建议做肾活检。肾检提示,光镜示系膜增殖性肾小球肾炎改变(重度);电镜示肾小球基底膜广泛变厚、劈裂。超超被诊断为Alport综合征。经过检查超超的父母和姐姐也有蛋白尿,也陆续通过基因检测诊断为同样的疾病。医生介绍,Alport综合征肾脏损害是一个逐渐进展的过程,依次是单纯血尿、中度蛋白尿、严重蛋白尿和肾功能下降,最终进入终末期肾脏病,即尿毒症。如果超超一家人的肾功能恶化到一定程度,将会面临透析和肾移植治疗。
三位一体
Alport综合征(AS)又称眼-耳-肾综合征,是由编码基底膜IV型胶原的基因突变(COL4An突变)所致的一种遗传性进行性肾小球基底膜疾病,累及肾脏,引起听力和眼部病变。
肾脏表现为血尿、蛋白尿、终末期肾病;听力改变表现为感音神经性耳聋:初为高频区听力下降,渐及全音域,甚至影响日常的对话交流;眼部病变可表现为前圆锥形晶状体,眼底黄斑周围点状和斑点状视网膜病变,视网膜赤道部视网膜病变表现为进行性近视,甚至导致前极性白内障或前囊自发穿孔。
其他症状有平滑肌瘤,肌发育不良,甲状腺疾病,AMME综合征(AS)伴精神发育迟缓、面中部发育不良及椭圆形红细胞增多症等)。
三个分类
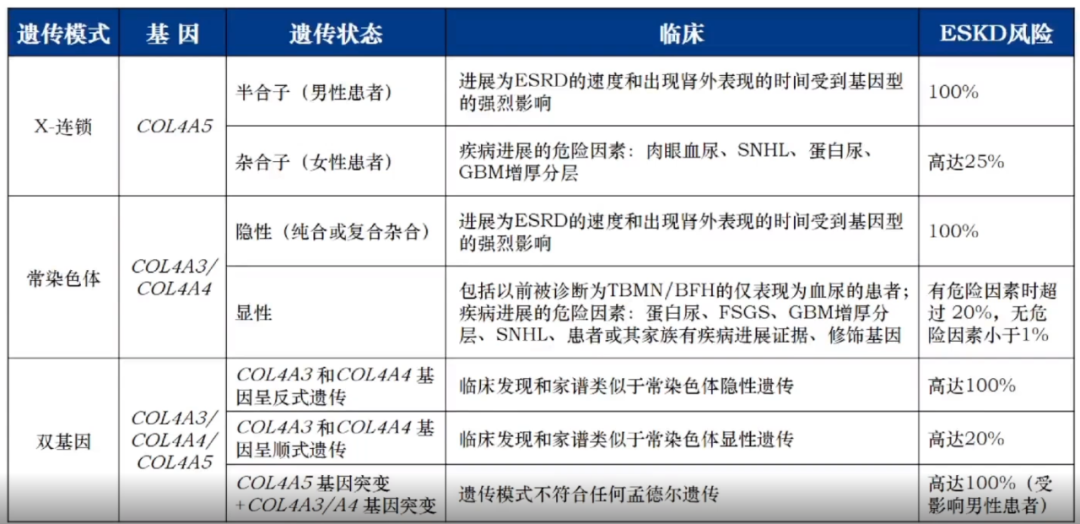
根据不同的遗传方式,AS可分为三种类型:
X连锁Alport综合征(XLAS)
常染色体隐性Alport综合征(ARAS)
常染色体显性Alport综合征(ADAS)
诊断
当持续性肾小球性血尿或血尿伴蛋白尿的患者,有以下任何一条应考虑AS额可能:
1.AS家族史
2.无明显其他原因的血尿、肾衰竭家族史
3.耳聋、圆锥形晶状体或黄斑周围斑点状视网膜病变
诊断依据
1.肾小球GBM Ⅳ型胶原α3、α4、α5链IF染色异常或皮肤EBM Ⅳ型胶原α5链IF染色异常
2.肾组织电镜示GBM致密层撕裂分层
3.COL4A5基因具有一个致病性变异或 COL4A3或者COL4A4基因具有两个致病性变异。
治疗
药物治疗
目前没有特异性的药物治疗,药物治疗的的目的在于控制尿蛋白,延缓病程的进展。
肾脏替代治疗
对于进展至终末期肾脏病者,肾移植是有效的治疗措施之一。Alport综合征患者肾移植后5年存活率在90%以上,10年存活率达70%以上。
基因治疗
近年来已确定了各种遗传型Alport综合征的突变基因,但基因治疗还存在一系列的问题,Alport综合征的基因治疗用于临床还需时日。
参考资料:
1. Alport综合征基因治疗进展 中华儿科杂志, 2024,62(7) : 692-695
2. 《Alport综合征诊治专家共识(2023版)》
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#Alport综合征# #眼-耳-肾综合征#
62