
Cell:能量代谢与免疫逃逸的双重挑战:YTHDF2如何塑造肿瘤生存策略?
2024-12-19 生物探索 生物探索 发表于陕西省

文章介绍 B 细胞恶性肿瘤治疗现状与问题,阐述 YTHDF2 在其中的双重作用机制(促进 ATP 合成和免疫逃逸),展示相关证据,介绍 YTHDF2 抑制剂 CCI - 38,展望 RNA 修饰研究对
引言
在过去的几十年里,B细胞恶性肿瘤(如急性淋巴细胞白血病 B-ALL 和B细胞淋巴瘤)已经成为威胁人类健康的重要疾病。这些恶性肿瘤通常起源于B细胞的异常增殖和分化,导致癌细胞快速生长并逃避免疫系统的监视。尽管近年来免疫疗法(如嵌合抗原受体T细胞疗法CAR-T和双特异性T细胞接合抗体BiTE)取得了显著进展,但治疗的长期效果仍然不尽如人意——约50%的患者在接受治疗后出现复发,其中抗原逃逸(Antigen Escape)是导致复发的主要原因之一。此外,B细胞恶性肿瘤的快速生长对能量供应提出了极高的需求,然而,这一过程中癌细胞如何维持高效的ATP合成与供能仍然是一个尚未完全揭示的关键科学问题。
在这一背景下,12月17日Cell的研究报道“YTHDF2 promotes ATP synthesis and immune evasion in B cell malignancies”,聚焦于YTHDF2这一重要的RNA修饰识别蛋白(Reader)。YTHDF2作为mRNA的m6A(N6-甲基腺嘌呤)和m5C(5-甲基胞嘧啶)双重识别因子,扮演着调控基因表达的关键角色。该研究发现,YTHDF2在B细胞恶性肿瘤中呈现异常高表达,并通过双重机制促进癌细胞的生存与免疫逃逸。一方面,YTHDF2通过识别m5C修饰的F型ATP合成酶亚基mRNAs,稳定其表达,显著提高ATP的合成效率,从而为癌细胞的快速生长提供充足的能量。另一方面,YTHDF2还能识别m6A修饰的CD19和MHC-II类分子mRNAs,促进其降解,削弱癌细胞对免疫系统的可见性,进而逃避免疫疗法的杀伤作用。
此外,研究人员通过筛选小分子化合物,发现了YTHDF2的抑制剂CCI-38,该药物能够显著抑制ATP合成,恢复CD19和MHC-II分子的表达,从而增强CAR-T细胞和BiTE疗法对肿瘤的杀伤效果。这一发现为B细胞恶性肿瘤的治疗提供了新的靶点和药物选择。
该研究首次揭示了YTHDF2在B细胞恶性肿瘤中的能量供给和免疫逃逸的双重作用机制,阐明了RNA修饰在肿瘤发生与免疫治疗中的重要作用,极大地拓宽了人们对B细胞恶性肿瘤的认知。YTHDF2抑制剂的成功应用不仅有望改善当前免疫疗法的疗效,也为未来肿瘤的精准治疗开辟了新方向。

B细胞恶性肿瘤:隐匿的“能量怪兽”
在血液系统肿瘤中,B细胞恶性肿瘤如急性淋巴细胞白血病(B-ALL)和B细胞淋巴瘤,是一类具有高度侵袭性且难以治愈的疾病。这些肿瘤起源于B细胞的异常增殖和分化,导致体内大量恶性B细胞积聚,不仅破坏正常免疫功能,还迅速扩散至骨髓、淋巴结等关键部位,威胁患者的生命。更令人棘手的是,B细胞恶性肿瘤具有极强的“适应性”,它们能够在免疫系统的压力下进化出逃避机制,特别是在接受免疫疗法后,部分癌细胞通过“抗原逃逸”机制隐藏自己,成为医生们难以消灭的“能量怪兽”。
近年来,以嵌合抗原受体T细胞疗法(CAR-T)和双特异性T细胞接合抗体(BiTE)为代表的免疫疗法为B细胞恶性肿瘤患者带来了新的希望。例如,CAR-T疗法通过改造T细胞,使其能够精准识别B细胞表面的CD19抗原并对癌细胞进行杀伤。然而,临床数据却显示,约50%的患者在治疗后出现复发,其中主要原因是癌细胞降低了CD19分子的表达,躲过了CAR-T的“追踪”,这一现象被称为抗原逃逸。
与此同时,B细胞恶性肿瘤的快速增殖对能量供应提出了前所未有的需求。研究发现,虽然恶性B细胞的线粒体数量较少,但它们却能够维持极高效的ATP合成,确保细胞迅速分裂和扩散。这种看似“违背生物常识”的现象表明,癌细胞必然存在一套独特的能量代谢机制,支撑其快速生长的“贪婪本性”。
因此,研究人员面临两大关键问题:第一,如何解决免疫疗法中癌细胞的抗原逃逸问题?第二,癌细胞是如何在有限的线粒体条件下维持高效的能量供应?揭开这些谜团,不仅能帮助人类更好地理解B细胞恶性肿瘤的生存策略,还将为新的治疗手段提供突破口。
RNA修饰与YTHDF2:肿瘤细胞的“幕后推手”
在基因表达的过程中,RNA分子曾被认为只是简单的“信息传递者”,负责将DNA编码的遗传信息转化为蛋白质。然而,近年来的研究揭示,RNA分子远比我们想象中复杂,它们的功能受到一系列化学修饰的精细调控。这些修饰,就像分子世界的“隐形墨水”,决定了RNA的稳定性、翻译效率以及在细胞内的去向。其中,最重要的两种修饰分别是m6A(N6-甲基腺嘌呤)和m5C(5-甲基胞嘧啶)。
m6A修饰是RNA分子上最常见的内部修饰之一,广泛存在于真核生物的mRNA中。它通过被“写入酶”(Writer)标记、被“擦除酶”(Eraser)去除,最终由“识别蛋白”(Reader)读取,调控RNA的降解和翻译。相比之下,m5C修饰虽然不如m6A普遍,但它同样影响mRNA的稳定性,尤其是在代谢调控等关键生物过程中发挥作用。
在这场RNA修饰的博弈中,YTHDF2成为了肿瘤细胞的“幕后推手”。YTHDF2是一种特殊的RNA修饰识别蛋白,能够同时识别m6A和m5C两种修饰,成为癌细胞快速增殖和免疫逃逸的核心工具。一方面,YTHDF2通过识别m5C修饰的F型ATP合成酶mRNAs,稳定其表达,促进ATP的高效合成,为癌细胞的快速分裂提供能量支持。另一方面,YTHDF2也能识别m6A修饰的CD19和MHC-II类分子mRNAs,促进它们的降解,帮助肿瘤细胞逃避免疫系统的追杀。
研究数据显示,YTHDF2在B细胞恶性肿瘤中异常高表达,其水平显著高于正常B细胞。这种表达的增加,使YTHDF2成为推动肿瘤发生、发展和治疗耐受的关键分子,就像一个隐藏在幕后却操控全局的“指挥官”。
正是YTHDF2对m6A和m5C“双重修饰”的精准读取,赋予了癌细胞强大的生存和适应能力。因此,靶向YTHDF2不仅有望切断癌细胞的“能量供给”,还能打破其“隐身术”,让肿瘤细胞再次暴露于免疫系统的火力之下。
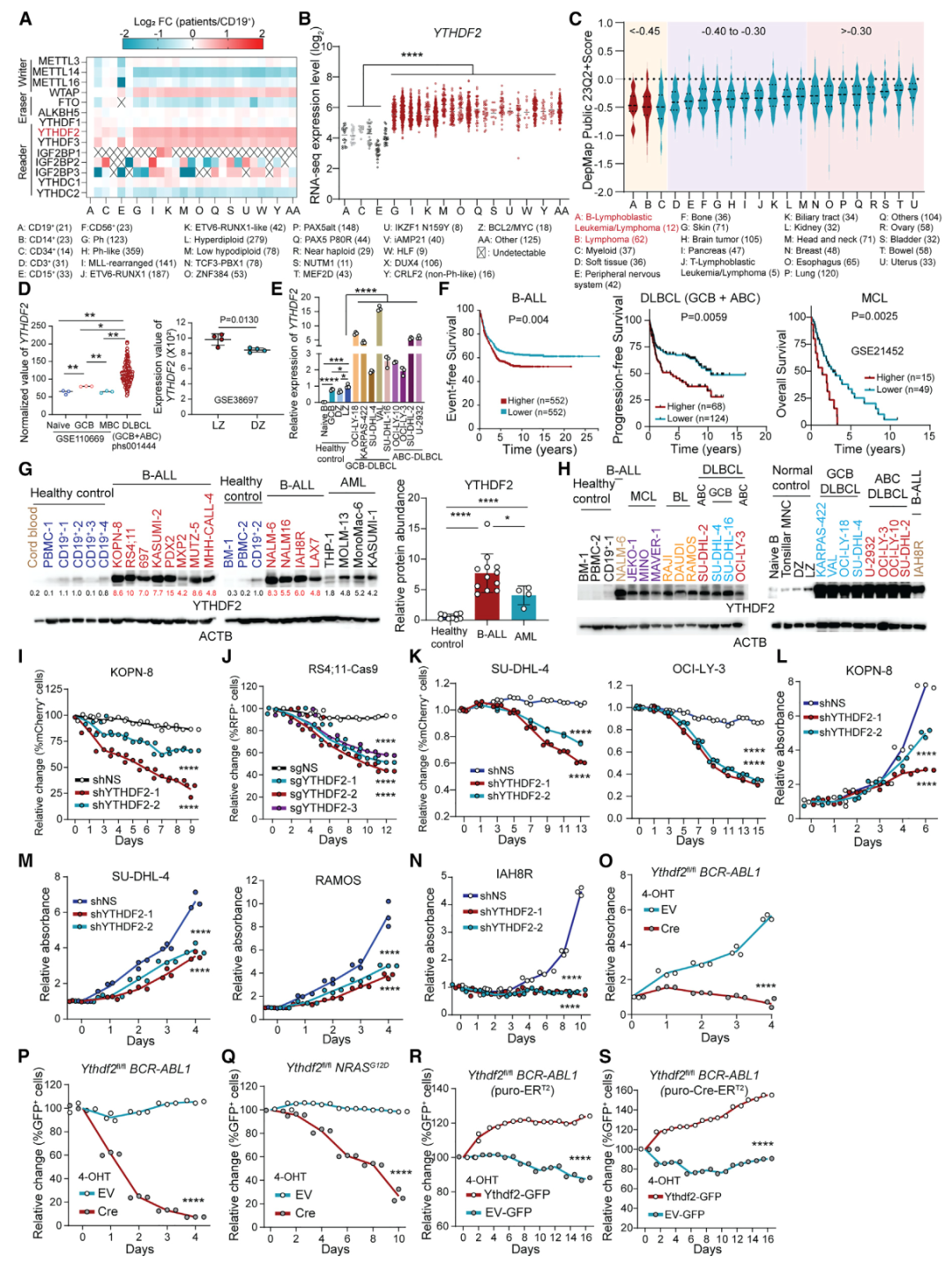
YTHDF2在B细胞恶性肿瘤中高表达并扮演促癌作用的证据(Credit: Cell)
(A) 热图分析(Heatmap)
通过对 B-ALL 患者的 RNA-seq 数据集分析,展示了与 m6A 修饰相关基因在肿瘤中的表达模式。结果显示,YTHDF2 在 B-ALL 患者中表达显著升高,并且在所有 m6A 机制基因中最显著。
(B) YTHDF2 表达水平
与 (A) 中的热图一致,分析了 YTHDF2 在患者中的丰度,进一步确认其在 B-ALL 患者中高度表达。
(C) 依赖性评分(DepMap 数据库)
通过 CRISPR-Cas9 基因敲除筛选数据,显示了 YTHDF2 对于 B 细胞白血病/淋巴瘤细胞的生存至关重要(依赖性评分显著降低),而对其他类型癌症的依赖性较低。这表明 YTHDF2 是 B 细胞恶性肿瘤中特异的关键基因。
(D) YTHDF2 mRNA 在 DLBCL 患者与健康对照中的丰度
RNA-seq 数据显示,YTHDF2 在弥漫性大 B 细胞淋巴瘤(DLBCL)患者中的表达显著高于健康对照组,进一步证实其在恶性 B 细胞中的高表达。
(E) 正常与恶性 B 细胞中 YTHDF2 的实时 qPCR 结果
实时定量 PCR 显示,YTHDF2 在多种恶性 B 细胞类型中的 mRNA 水平显著高于正常 B 细胞,数据支持其促癌基因的特性。
(F) 生存分析
基于不同 YTHDF2 表达水平,将 B-ALL、DLBCL 和 MCL(套细胞淋巴瘤)患者分组进行生存分析。高表达 YTHDF2 的患者表现出较短的总体生存期(OS)、无进展生存期(PFS)或无事件生存期(EFS),说明其高表达与不良预后密切相关。
(G) 白血病与健康细胞中的 YTHDF2 蛋白表达
利用统计分析比较,显示YTHDF2 在白血病细胞中的蛋白表达显著高于健康细胞,进一步支持其作为促癌因子的作用。
(H) 正常与恶性 B 细胞中的 YTHDF2 蛋白表达
进一步验证了YTHDF2 蛋白在恶性 B 细胞中显著高于正常 B 细胞,结果与 RNA 水平一致。
(I–K) YTHDF2 敲低对恶性 B 细胞竞争力的影响
通过在不同 B 细胞恶性肿瘤模型中敲低 YTHDF2(使用 mCherry+-shRNA 或 RFP+-sgRNA 标记),发现 YTHDF2 敲低显著降低了肿瘤细胞的竞争能力,说明其在维持肿瘤细胞生存中的重要作用。
(L–O) 敲低 YTHDF2 对细胞生长和增殖的影响
通过对 B-ALL、淋巴瘤细胞和患者衍生的异种移植(PDX)模型细胞敲低 YTHDF2,发现这些细胞的生长和增殖能力显著下降。特别是在小鼠模型中,使用 Ythdf2 条件性敲除(cKO)也表现出类似的抑制效果。
(P–S) YTHDF2 过表达对恶性 B 细胞的促进作用
通过在转染了空载体-GFP 或 Ythdf2-GFP 的 B-ALL 细胞中进行竞争力和增殖实验,发现过表达 YTHDF2 可显著增强恶性 B 细胞的竞争力和增殖能力,进一步证明其作为促癌基因的功能。
YTHDF2如何助力癌细胞“充能”:揭开ATP高效合成的秘密
癌细胞的快速生长和增殖,就像一座永不停歇的“工厂”,需要源源不断的能量作为支撑。而这种能量的核心来源,正是细胞线粒体中通过氧化磷酸化(OXPHOS)产生的三磷酸腺苷(ATP)。然而,B细胞恶性肿瘤面临一个天然的“能量瓶颈”:它们分裂速度极快,但线粒体数量却远低于其他类型的癌细胞。这让研究人员不禁好奇,它们是如何在“缺乏燃料仓库”的情况下,实现高效能量供应的呢?答案的关键,就隐藏在YTHDF2和RNA修饰的巧妙配合中。
研究发现,YTHDF2能够识别并结合m5C修饰的特定mRNAs,尤其是负责ATP合成的F型ATP合成酶亚基(ATP5PB、ATP5MF和ATP5MG)mRNAs。通过这一结合,YTHDF2能够稳定这些mRNAs,防止它们被降解,从而提高ATP合成酶亚基的蛋白质表达水平。ATP合成酶作为线粒体中“能量制造”的核心机器,加速了ATP的生成,为癌细胞提供了充足的能量供应。
实验数据显示,当YTHDF2被敲低时,癌细胞的氧气消耗率(OCR)和ATP合成显著下降,细胞增殖也随之受阻。这一现象在多种B细胞恶性肿瘤中得到验证,包括急性淋巴细胞白血病(B-ALL)和弥漫性大B细胞淋巴瘤(DLBCL)。相反,过表达YTHDF2或其m6A结合突变体(m6A-MUT)时,ATP合成效率迅速提升,癌细胞的能量供应得到极大保障,推动其快速增殖和扩散。
更令人惊讶的是,YTHDF2的这一功能完全依赖于其对m5C修饰的特异识别,而非m6A修饰。这揭示了YTHDF2在癌细胞中的一种全新机制:通过稳定m5C修饰的ATP合成酶亚基mRNAs,它巧妙地维持了癌细胞的“能量充足状态”,让这些肿瘤细胞在能量短缺的逆境中仍能高速运转,肆意扩张。
“隐身术”揭秘:YTHDF2如何帮助癌细胞逃避免疫打击
肿瘤细胞与免疫系统之间的斗争,就像一场持续不断的“猫鼠游戏”。免疫疗法,如CAR-T细胞疗法,通过精准识别癌细胞表面的CD19抗原,发挥强大的杀伤作用。然而,令人沮丧的是,部分癌细胞似乎掌握了“隐身术”,在接受治疗后,表面CD19分子的表达逐渐消失,导致免疫疗法失效。这一现象背后的“幕后黑手”,正是YTHDF2。
研究发现,YTHDF2能够识别m6A修饰的mRNAs,并通过促进其降解,调控特定基因的表达。在B细胞恶性肿瘤中,YTHDF2直接作用于CD19和MHC-II类分子(如HLA-DMA和HLA-DMB)的mRNAs,导致它们快速降解,显著降低了这些关键抗原分子的表达水平。CD19是CAR-T疗法的主要靶点,而MHC-II类分子则在抗原呈递中发挥重要作用,它们的减少使癌细胞有效“隐藏”在免疫系统的视野之外,从而实现免疫逃逸。
实验数据显示,敲低YTHDF2后,CD19和MHC-II类分子的mRNA稳定性显著增强,蛋白表达水平明显提高,癌细胞表面的抗原呈递能力恢复,免疫细胞对癌细胞的杀伤效果大幅提升。相反,过表达YTHDF2会进一步降低CD19和MHC-II分子的表达,使肿瘤细胞更难被免疫系统识别。这一机制在CAR-T细胞与双特异性抗体(BiTE)疗法中尤为关键,因为CD19的丢失正是导致治疗失败和肿瘤复发的主要原因之一。
更有趣的是,YTHDF2识别m6A修饰的CD19和HLA-DMA/B mRNAs,不依赖于其m5C修饰结合能力,表明YTHDF2在能量代谢与免疫逃逸之间发挥“双重作用”,为肿瘤细胞提供了“生存武器”。这一特性让YTHDF2成为B细胞肿瘤的核心驱动因素之一,也是目前治疗耐药与复发问题的重要突破口。
突破困境:YTHDF2抑制剂CCI-38的横空出世
面对YTHDF2这一癌细胞“能量供给”和“免疫逃逸”的核心帮凶,研究人员开始通过高通量化合物筛选,寻找能够特异性抑制YTHDF2的小分子药物。
通过基于结构的虚拟筛选技术,从79种潜在候选化合物中锁定了CCI-38,并进一步通过药物亲和性反应目标稳定性试验(DARTS)和荧光蛋白热稳定性实验(FTSA)确认其对YTHDF2的高特异性。CCI-38能够与YTHDF2的RNA结合口袋相互作用,阻断YTHDF2与m6A或m5C修饰mRNAs的结合。这一机制使得CCI-38在分子层面有效瓦解了YTHDF2的功能。
在功能实验中,CCI-38展现了令人惊叹的双重抑制效果:一方面,它抑制了YTHDF2对m5C修饰ATP合成酶亚基mRNAs的稳定作用,显著降低ATP合成,直接削弱了癌细胞的“能量供应链”;另一方面,CCI-38阻断了YTHDF2对m6A修饰的CD19和MHC-II分子mRNAs的降解作用,恢复了肿瘤细胞的抗原表达,使其重新暴露于免疫系统的攻击之下。
此外,CCI-38不仅在实验室细胞系中表现出强大的抑瘤作用,还在动物模型中展现了极佳的治疗效果。研究发现,CCI-38能够显著减少肿瘤负荷,延长移植小鼠的生存期;与CAR-T细胞疗法联合使用时,更是表现出强大的协同作用,大幅提高了CAR-T细胞对肿瘤细胞的杀伤效率。
实验室到临床:CCI-38如何帮助CAR-T疗法“重振旗鼓”
研究团队通过一系列实验,证明CCI-38不仅能够单独抑制肿瘤进展,还能与CAR-T疗法协同作用,显著提升免疫疗法的效果。在动物模型中,研究人员将CCI-38应用于移植了复发性B细胞恶性肿瘤的小鼠(这些肿瘤对CAR-T疗法表现出耐受性)。结果显示,CCI-38能够有效恢复CD19和MHC-II分子的表达,使癌细胞重新暴露在CAR-T细胞的杀伤范围内。相比仅使用CAR-T疗法的对照组,联合CCI-38的小鼠群体肿瘤负荷明显减少,生存期显著延长。
在另一项实验中,研究团队利用从患者样本中提取的肿瘤细胞,进一步验证了CCI-38的临床潜力。他们从一位因抗原逃逸复发的B-ALL患者身上分离出CD19低表达的肿瘤细胞,并将其用于体外实验。结果表明,CCI-38治疗能够将CD19表达恢复至初诊时的水平(甚至更高),显著增强了CAR-T细胞对这些肿瘤细胞的杀伤能力。这种恢复效果在其他患者样本中也得到了验证,显示出CCI-38的广泛适用性。
更令人鼓舞的是,联合治疗不仅增强了CAR-T的杀伤效果,还显示出强大的协同抗肿瘤作用。在CAR-T细胞无法完全清除肿瘤的情况下,CCI-38通过减少癌细胞的能量供应和免疫逃逸,为CAR-T疗法争取了“第二次机会”。这种双重机制的发挥,极大地提高了免疫疗法的总体成功率。
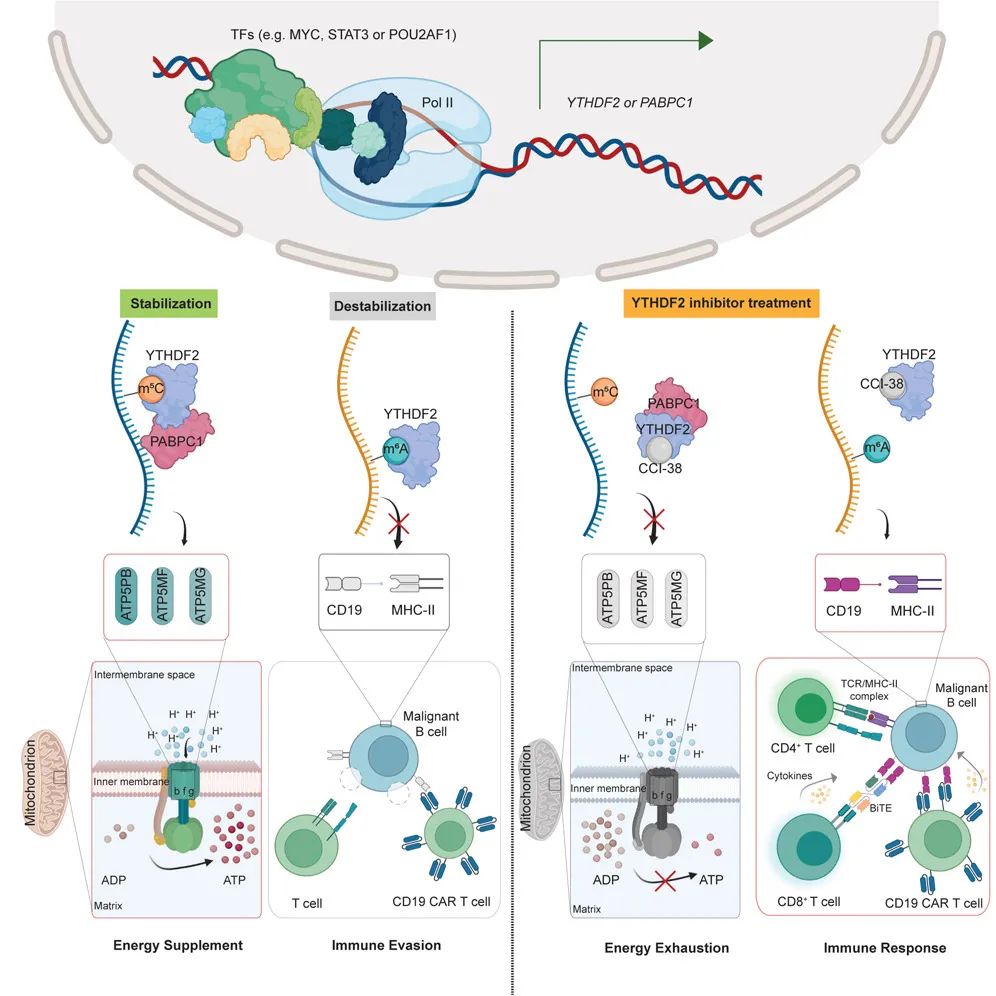
模式图(Credit: Cell)
RNA修饰研究如何改变肿瘤治疗格局?
癌症的研究历史已经证明,突破性的科学发现往往来自对看似普通现象的深入挖掘。而RNA修饰,作为生命科学领域的一颗新星,正逐渐揭开其在基因表达调控中的神秘面纱。以YTHDF2为代表的RNA修饰识别蛋白,已经成为研究人员关注的焦点。这些分子不仅帮助我们重新理解肿瘤的发生机制,还为未来肿瘤的精准治疗提供了全新的靶点。
该研究展示了YTHDF2如何通过识别m6A和m5C修饰,为B细胞恶性肿瘤提供“双重助力”:一方面,YTHDF2稳定m5C修饰的ATP合成酶亚基mRNAs,维持肿瘤的高效能量供应;另一方面,它促进m6A修饰的CD19和MHC-II分子的降解,帮助癌细胞实现免疫逃逸。这种“双面角色”使YTHDF2成为B细胞肿瘤发展的关键推动因素,也为靶向YTHDF2的药物开发提供了明确方向。通过该研究开发的CCI-38抑制剂,研究人员成功抑制了YTHDF2的功能,不仅切断了癌细胞的“能量生命线”,还恢复了免疫系统对肿瘤细胞的精准打击能力。
更重要的是,YTHDF2只是RNA修饰调控网络中的冰山一角。研究表明,其他m6A或m5C相关蛋白同样可能在肿瘤发生、发展和治疗耐药中发挥重要作用。这意味着,RNA修饰领域蕴藏着无数潜在的治疗靶点,有望为肿瘤患者带来更多突破性疗法。例如,未来的研究可以探索RNA修饰如何调控肿瘤微环境,或研究RNA修饰的可逆性在抗癌药物开发中的应用潜力。
此外,该研究还展现了基础科学如何为临床实践提供直接的指导。从RNA修饰的分子机制解析,到CCI-38的开发,再到联合CAR-T疗法的显著效果,他们完成了一次从实验室到临床的创新之旅。RNA修饰研究不仅揭示了癌症的复杂性,也为我们提供了重新设计抗癌策略的工具箱。
展望未来,随着RNA修饰调控机制的深入研究,以及更多靶向药物的成功开发,肿瘤的精准治疗将迎来全新格局。RNA修饰的故事才刚刚开始,而它的潜力,足以让我们对癌症的治愈充满希望。这一领域的持续探索,或许会成为改变对抗癌症命运的决定性力量。
参考文献
https://www.cell.com/cell/abstract/S0092-8674(24)01324-2
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#免疫逃逸# #B细胞恶性肿瘤#
8