遗传性痉挛性截瘫:伴耳聋和无精子症|病例分享
2023-07-04 神经科学论坛 神经科学论坛 发表于上海

在这项研究中,我们报道一名AP5Z1基因大量缺失的43岁中国男性患者的临床特征和影像学表现。该患者报告了痉挛性截瘫、听力障碍和不孕症。
论坛导读:遗传性痉挛性截瘫(Hereditary spastic paraplegia,HSP)是一种罕见的神经退行性疾病,以进行性下肢痉挛和无力为特征。HSP可通过常染色体显性、常染色体隐性、X连锁隐性或线粒体方式遗传。已经鉴定了几种基因型(从SPG1到SPG82)和致病基因,最常见的致病基因是SPAST、ATL1和REEP1。AP5Z1中的破坏性变异与SPG48相关,spg 48是一种罕见的常染色体隐性疾病,具有多样化的临床表型。
迄今为止,我们知道14例与该基因相关的病例报告。在这项研究中,我们报道一名AP5Z1基因大量缺失的43岁中国男性患者的临床特征和影像学表现。该患者报告了痉挛性截瘫、听力障碍和不孕症。

病例资料
53岁男性,因进行性行走困难和下肢远端无力而入院。他在小学期间成绩很低,后来没有完成学业。他28岁结婚。当他35岁的时候,他被诊断患有不育症。2010年,患者在行走过程中出现不稳定和轻微摇晃。行走不稳定性在2015年恶化,当时患者报告从蹲姿站起来有困难。2019年,患者开始使用拐杖行走。从2017年开始,双耳听力逐渐下降。自2019年以来,患者只识别大声说出的声音,耳朵没有疼痛、感染和/或耳鸣。患者的父母是堂兄妹,家族史没有明显的相似的神经和听觉表现。
在检查中,我们观察到双侧痉挛性步态、腱反射亢进、踝阵挛(+)和双侧锥体束征。龙伯格测试呈阳性。轻度痉挛性构音障碍也很明显。感觉检查显示大脚趾失去振动感,下肢针刺感减少。痉挛性截瘫评定量表(SPRS)得了19分。空间和时间方向正常,但注意力和计算能力受损。听力损失影响双耳,左(+)和右( )铃音测试呈阳性。韦伯测试偏向右耳。睾丸的外观和大小都正常。血液实验室检查和脑脊液评估正常,包括全身激素。
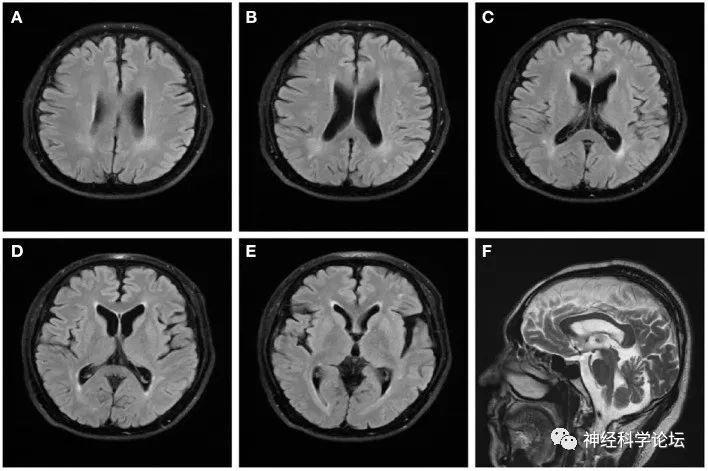
脑磁共振成像(MRI)显示轻度脑萎缩,在液体衰减反转恢复(FLAIR)序列上表现为脑室周围白质高信号。颈椎和腰椎MRI描述了腰椎的退行性改变和多个椎间盘的轻度扩张,没有明显的脊髓压迫或其他异常信号。肌电图检查显示右侧腓总神经和左侧胫神经的复合肌肉动作电位(CMAPs)振幅降低。在双侧腓肠神经中观察到感觉神经动作电位(SNAPs)的潜伏期延长,在双侧正中神经和胫神经中F波的潜伏期增加。双侧胫骨H反射检查显示潜伏期延长,胫骨前肌出现神经源性损伤。听力评估发现左侧为感音神经性耳聋,右侧为混合性耳聋。诱发电位提示双耳听觉阈值增加。在脑干听觉诱发电位上,我们发现了双侧延长的ⅰ波和ⅲ波。体感诱发皮层电位显示左侧P40潜伏期延长。认知测试的结果是简易精神状态测验(MMSE)27分,蒙特利尔认知评估18分。
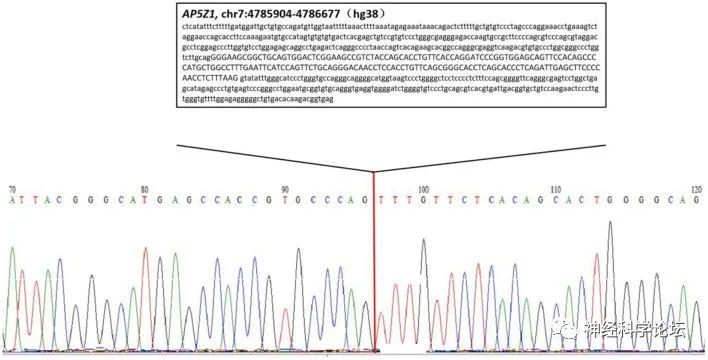

我们通过Sanger测序确定了基因缺失的位置,确定了涉及AP5Z1基因外显子10的chr7:4785904-4786677区域的纯合缺失,NM_014855.3,c.1133-345_1311 + 249del,p.G378Vfs*93X。使用软件Mutalyzer 2.0.35,我们发现该缺失导致氨基酸合成的提前终止。患者的兄弟无症状,通过荧光定量聚合酶链反应(PCR)鉴定出杂合子突变。引物如下:AP5Z1-10(正向:AACCAGTCACAGAAGCACGG,反向:GAACAGGTGGAGGTTGTCCC),PCR产物的序列用ABI 9700遗传分析仪(ABI,加利福尼亚州福斯特市)测定。反应系统:总体积10ul,包括1ul基因组DNA模板,5ul SYBR Green Mix,0.5ul上游和下游引物,3ul ddH2O。反应条件:95℃,30秒,(95℃,15秒;60°C,15s72°C,15秒)× 46个循环。最终数据通过2δCt进行分析。结果显示,相对于健康人的两个拷贝,病人有0个拷贝,他的兄弟有一个拷贝。根据美国医学遗传学和基因组学学院的标准和指南,这种缺失突变被认为是一种致病性变异。基于临床特征、影像学表现和基因异常,我们诊断该患者为SPG48。口服巴氯芬和替扎尼定改善了他的症状。
延伸阅读
2010年,Slabicki等人发现SPG48是一种与HSP相关的新基因型。SPG48可以被定义为一种继发性溶酶体贮积病,涉及衔接蛋白(APs),从AP1到AP5范围内普遍表达的蛋白复合物。AP5是一种阳离子非依赖性甘露糖6-磷酸受体,位于内体和溶酶体中,将高尔基膜蛋白1转运至高尔基体。AP5在非神经元和神经元组织中表达,如大脑皮层、海马和小脑。神经元细胞对溶酶体功能障碍和蛋白质积累异常敏感。亚型SPG48与AP5的ζ亚单位突变有关,削弱了AP5的复合物形成。这进而导致溶酶体结构和功能的缺陷。SPG48的小鼠模型显示,功能丧失的AP5变体阻断了自噬,导致自噬空泡的异常积聚和轴突变性。双等位基因AP5Z1突变患者的尸检结果显示,包括基底神经节、脑干和脊髓在内的皮质和皮质下区域广泛变性。这种改变在神经退行性疾病中很常见,包括HSP、肌萎缩性侧索硬化和脊髓小脑共济失调。到今天为止,全世界已经报告了14例SPG48病例。其中大多数以点突变为特征,而我们的患者报告了AP5Z1基因的片段性缺失。总的来说,SPG48的发病年龄跨越数十年,临床表现高度异质性,包括痉挛性截瘫、尿失禁、共济失调、智力残疾、感觉运动神经病、帕金森综合征、肌张力障碍和眼部障碍[包括色素性视网膜病、视神经萎缩、白内障、青光眼和眼肌麻痹,非典型SPG48甚至没有表现出痉挛性截瘫。在大多数患者中,脑部MRI显示放射冠、半卵圆中心和侧脑室周围的白质病变。“猞猁之耳”的影像学征象表明存在基因突变,这可能是过敏性紫癜的特征。胼胝体变窄是这些患者的另一个相关的影像学特征。少数患者脑部MRI显示正常,而个别病例报告了弥漫性脑萎缩和/或异常脊髓信号。
我们的患者报告了痉挛性截瘫、周围神经病和轻度认知障碍,并首次发现无精子症和严重的双侧听力损失。先前的研究发现HSP神经元的线粒体长度和密度显著减少,表明异常的线粒体形态可能导致轴突缺陷。最近的一项研究发现,受损的线粒体动力学导致SPG11和SPG48神经元的轴突变性,导致轴突效率降低和缩短。线粒体功能也与人类精子活力和形态有关。此外,线粒体功能的丧失与耳聋有关。因此,我们可以推测AP5Z1基因突变可能影响精子发生和听力。SPG48极大地影响中枢神经系统,具有复杂多样的临床和影像学特征。临床医生应注意疑似SGP48病例的非神经系统表现,因为疾病的早期识别将减少不必要的评估和治疗。目前对SPG48尚无有效的治疗方法,包括巴氯芬和替扎尼定在内的对症支持治疗可中度改善痉挛性截瘫和尿失禁症状,未来的疗法可能会恢复线粒体功能。
罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
全文索引:
Jin P, Wang Y, Nian N, Wang GQ, Fu XM. Hereditary spastic paraplegia (SPG 48) with deafness and azoospermia: A case report. Front Neurol. 2023 Apr 3;14:1156100. doi: 10.3389/fneur.2023.1156100. PMID: 37077568; PMCID: PMC10106626.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言